筆記目錄
本文是臨床評估主題的第一篇筆記,主要整理了以下指引與法規:
- Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019)
- Clinical Evaluation (MEDDEV 2.7/1,Rev. 4)
- Medical Device Regulation (EU) 2017/745
| 後續新增了澳洲 TGA〈Clinical evidence guidelines for medical devices〉(V3.0 Nov 2021,下稱「TGA 指引」) 部分資訊。 |
希望未來可以整理出下面的主題:
- Software as a Medical Device (SAMD) 的臨床評估方式
- 臨床評估的類似品 (Equivalence,aka Comparable devices) 判定方式
| 想了解「資料識別、評讀與分析的實作方法」嗎?請看「再談臨床評估之資料識別 (Identification)、評讀 (Appraisal) 與分析 (Analysis)」。 |
1. 什麼是 Clinical evaluation?
拿 MDR (2017/745) 的定義來說:
‘Clinical evaluation’ means a systematic and planned process to continuously generate, collect, analyse and assess the clinical data pertaining to a device in order to verify the safety and performance, including clinical benefits, of the device when used as intended by the manufacturer.
「臨床評估」是一個事前規劃好的系統化程序,可以持續地產生、收集、分析與評估與目標醫材相關的臨床資料,以驗證目標醫材於預期使用上的安全、效能與臨床效益。
Medical Device Regulation (EU) 2017/745 | (44) of Article 2 Definitions
針對上文提到的「Continuously」(持續地),下面進一步解釋「臨床評估應在『整個器械的生命週期』中更新」:
The clinical evaluation and its documentation shall be updated throughout the life cycle of the device concerned with clinical data obtained from the implementation of the manufacturer’s PMCF plan in accordance with….
Medical Device Regulation (EU) 2017/745 | Sec. 11 of Article 61 (Clinical Evaluation)
另外,IMDRF 指引也有類似定義:
Clinical evaluation is a set of ongoing activities that use scientifically sound methods for the assessment and analysis of clinical data to verify the safety, clinical performance and/or effectiveness of the medical device when used as intended by the manufacturer.
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | 1.0 Introduction
| 上段 IMDRF 中提到的「Clinical performance」是什麼? 節錄來自 EN ISO 14155:2011 (被 MEDDEV 2.7/1 Rev. 4 引用) 的定義: “behaviour of a medical device or response of the subject(s) to that medical device in relation to its intended use, when correctly applied to appropriate subject(s).” |
2. 何時做 Clinical evaluation?
2.1 首次執行時機
Clinical evaluation is an ongoing process conducted throughout the life cycle of a medical device. It is first performed during the development of a medical device in order to identify data that need to be generated for regulatory purposes and will inform if a new device clinical investigation is necessary, together with the outcomes which need to be studied. It is then repeated periodically as new safety, clinical performance and/or effectiveness information about the medical device is obtained during its use.
臨床評估是一個貫穿器材生命週期、持續進行的程序。首次執行時機是在研發階段,目的是為了 1) 識別法規所須的資料,以及 2) 了解是否有進行臨床試驗的需求。
在之後此器械實際使用過程中,當有新的安全、臨床效能、和/或功效資訊時,臨床評估須定期地重覆執行。
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | 1.0 Introduction
另外,有更簡短的說明:
Clinical evaluation is conducted throughout the life cycle of a medical device, as an ongoing process.
臨床評估作為一個持續進行的程序,在整個器材的生命週期中都會進行。
Usually, it is first performed during the development of a medical device in order to identify data that need to be generated for market access. Clinical evaluation is mandatory for initial CE-marking and it must be actively updated thereafter.
一般來說,首次執行是在器材的研發階段,目的是為了識別市場准入所須要的資料。
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 6.2. When is clinical evaluation undertaken and why is it important?
2.2 產品上市後更新時機
The clinical evaluation and its documentation shall be updated throughout the life cycle of the device concerned with clinical data obtained from the implementation of the manufacturer’s PMCF plan in accordance with Part B of Annex XIV and the post-market surveillance plan referred to in Article 84.
臨床評估與其相關文件,應在其完整生命週期內,利用執行 PMCF plan 與 PMS plan 索取的臨床資料持續更新。
For class III devices and implantable devices, the PMCF evaluation report and, if indicated, the summary of safety and clinical performance referred to in Article 32 shall be updated at least annually with such data.
至於 Class III devices 和 Implantable devices,每年應至少利用臨床評估等相關資料更新一次 PMCF evaluation report 和 SSCP。
Medical Device Regulation (EU) 2017/745 | Sec. 11 of Article 61 (Clinical Evaluation)
若非上述 MDR 規定的更新週期,一般來說,當 PMS 發現會影響產品的
- Risk/benefit profile、
- Clinical performance、和
- Clinical safety 時,
要及時更新臨床評估 (Required to be fed into the clinical evaluation process in a timely manner) (Sec 6.2.3. of MEDDEV 2.7/1 (Rev. 4)。
| 有關 Risk/benefit profile、Benefit-risk ratio、Benefit/risk determination 等等的利益與風險評估,請參考另篇筆記「醫療器材上市前審查考量之利益與風險權衡要素 (Benefit-Risk evaluation)」。 |
| 雖然臨床評估需要來自 PMS 的資訊,但它同時也會提供 PMS 和 Risk management process 新資訊。也就是說,可能因為臨床評估,而要變更風險管理報告、使用說明書、PMS 等。(Sec. 6.2.3.b. of MEDDEV 2.7/1 (Rev. 4)) |
3. 為什麼要 Clinical evaluation?
應該大部分公司都是因為「法規要求」才做臨床評估的吧!
但法規哪裡要求了呢?
(最討厭整天說「法規要求」卻找不出法源依據,或等被問「哪條法規要求了呢?」才開始找依據的人了!在說「因為法規要求而必須做 xx」之前,應該要先確定有這要求吧。)
以歐盟醫療器材法規 (MDR) 來說,在 Article 10 (General obligations of manufacturers) 要求製造廠要執行臨床評估:
Manufacturers shall conduct a clinical evaluation in accordance with the requirements set out in Article 61 and Annex XIV, including a PMCF.
The quality management system shall address at least the following aspects:…Clinical evaluation in accordance with Article 61 and Annex XIV, including PMCF.
| “For all class III devices and for the class IIb devices referred to in point (b) of Article 54(1), the manufacturer may, prior to its clinical evaluation and/or investigation, consult an expert panel as referred to in Article 106…” (Sec. 2 of Article 61 of MDR) 依上述規定,MDR 針對 Class III implantable devices 與部分 Class IIb active devices 的臨床評估特別要求「Clinical evaluation consultation procedure」,要在評估前先諮詢 Expert panels (專家小組)。 詳情請參考 MDR 的 Article 54 (Clinical evaluation consultation procedure for certain class III and class IIb devices)。 但奇怪的是,MDR Article 61 的 Sec.2 末段說:「The manufacturer may not invoke any rights to the views expressed by the expert panel with regard to any future conformity assessment procedure.」 也就是說,若未來在歐盟符合性評鑑過程中被挑出問題,製造廠不可以說:「啊當初有問專家小組,他們這樣說的啊!」(所以…專家小組可以隨便嘴砲喔?) |
至於美國,則設計管制指引中提到相關要求:
VALIDATION METHODS. Many medical devices do not require clinical trials. However, all devices require clinical evaluation and should be tested in the actual or simulated use environment as a part of validation.
許多醫材不一定需要臨床試驗,但所有醫材都須要臨床評估。
Design Control Guidance for Medical Device Manufacturers (1997) | II. DEFINITIONS
且在美國 FDA 的品質管理系統稽核指引內,也有提醒稽核員須確認製造廠是否有完成相關要求:
Design validation involves the performance of clinical evaluations and includes testing under actual or simulated use conditions. Clinical evaluations can include clinical investigations or clinical trials, but they may only involve other activities. These may include evaluations in clinical or non-clinical settings, provision of historical evidence that similar designs are clinically safe, or a review of scientific literature.
Guide to Inspections of Quality Systems (QSIT, 1999) | Design Controls
| 因為 SaMD (醫療器材軟體) 的特殊性,IMDRF 特別制訂〈Software as a Medical Device (SAMD): Clinical Evaluation〉(WG/N41FINAL:2017),美國 FDA 也有採認此指引。 另外,歐盟 MDCG 也有相關指引〈Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software〉(MDCG 2020-1,2020)。 關於這個,我們之後會專門整理一篇來分享。 |
那為何歐美要規定製造廠做臨床評估這種麻煩事勒?當然是為了保護使用者。
MDR 解釋如下:
Confirmation of conformity with relevant general safety and performance requirements set out in Annex I under the normal conditions of the intended use of the device, and the evaluation of the undesirable side-effects and of the acceptability of the benefit-risk ratio referred to in Sections 1 and 8 of Annex I, shall be based on clinical data providing sufficient clinical evidence, including where applicable relevant data as referred to in Annex III.
1) 確認器材在預期用途的正常條件下,可符合相關 GSPR;
2) 評估不良副作用,和
3) 利益風險比可接受度時,
應根據臨床資料所提供的充分臨床證據。
Medical Device Regulation (EU) 2017/745 | Sec. 1 of Article 61 (Clinical Evaluation)
另外,IMDRF 指引也有說明臨床評估的目的:
The evaluation must also address any clinical claims made about the device, the adequacy of product labelling and product information (particularly contraindications, precautions/warnings), and the suitability of instructions for use.
Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019) | 5.0 General principles of clinical evaluation
A key goal of the clinical evaluation is to establish that any risks associated with the use of the medical device are acceptable when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety.
臨床評估的關鍵目標是,
1) 確認和使用醫材相關的任何風險,在與病人利益相權衡後是可接受的,並且
2) 符合對健康與安全的高度保護。
Clinical Evidence – Key Definitions and Concepts (IMDRF MDCE WG/N55 FINAL:2019) | 4.3 Clinical evaluation
4. Clinical data & Clinical evidence 是什麼?
「什麼是 Clinical evaluation」已於上一章節討論過,但 Clinical data、Clinical evidence 又是什麼呢?
4.1 Clinical data 是什麼?
‘Clinical data’ means information concerning safety or performance that is generated from the use of a device and is sourced from the following:
– clinical investigation(s) of the device concerned (目標醫材的臨床試驗),
– clinical investigation(s) or other studies reported in scientific literature, of a device for which equivalence to the device in question can be demonstrated (類似品的臨床試驗,或其它科學文獻),
– reports published in peer reviewed scientific literature on other clinical experience of either the device in question or a device for which equivalence to the device in question can be demonstrated (目標醫材或類似品的臨床使用經驗的相關科學文獻,且此文獻須經同儕審查),
– clinically relevant information coming from post-market surveillance, in particular the post-market clinical follow-up (上市後監督,尤其是上市後臨床追蹤的臨床相關資訊).
Medical Device Regulation (EU) 2017/745 | (48) of Article 2 Definitions
| 上面第三點內的「Clinical experience」是什麼呢? 在 IMDRF 的〈Clinical Evaluation〉(WG/N56FINAL:2019) 說明如下: “These types of clinical data are generated through clinical use that is outside the conduct of clinical investigations and may relate to either the device in question or comparable devices. Such types of data may include: – post market surveillance reports, registries or medical records (which may contain unpublished long term safety, clinical performance and/or effectiveness data); – adverse events databases (held by either the manufacturer or regulatory authorities); and – details of clinically relevant field corrective actions (e.g. recalls, notifications, hazard alerts).” 要特別注意的是,MDR 規定 Clinical experience 的相關文獻,須經同儕審查才可用於臨床評估,比 IMDRF 上面的要求高! 後面「7.2 Clinical experience」將進一步討論。 |
4.2 Clinical evidence 是什麼?
‘Clinical evidence’ means clinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer.
「臨床證據」是指,和目標醫材相關的臨床資料與臨床評估結果,且具有足夠的資料量與品質,可用來評估目標醫材依製造廠預期來使用時是否安全,以及是否有達到預期臨床效益。
Medical Device Regulation (EU) 2017/745 | (51) of Article 2 Definitions
| 如何知道是否有「足夠」的資料量和品質呢?這應該看似又是主觀判斷,但可參考下面解釋: 「Clinical evaluation of medical devices that are based on existing, established technologies and intended for an established use of the technology is most likely to rely on compliance with recognised standards and/or literature review and/or clinical experience of comparable devices. High risk devices, those based on technologies where there is little or no experience, and those that extend the intended purpose of an existing technology (i.e. a new clinical use) are most likely to require clinical investigation data. The manufacturer will need to give consideration to the advantages and limitations of each data type.」 – Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019) | What is the scope of a clinical evaluation? 上面這段話,也已筆記於「6.4 資料來源與資料型態」。 |
4.3 Clinical data / evaluation / evidence 三者關係為何?
The clinical evaluation report and the relevant clinical data constitute the clinical evidence for conformity assessment.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 6.3. How is a clinical evaluation performed?
上段話可簡化成「Clinical data + Clinical evaluation report = Clinical evidence」。
換句話說,三者的關係是:Clinical data 經 Clinical evaluation 評估分析後,產出可證明目標醫材安全,且達到預期臨床效益的 Clinical evidence。
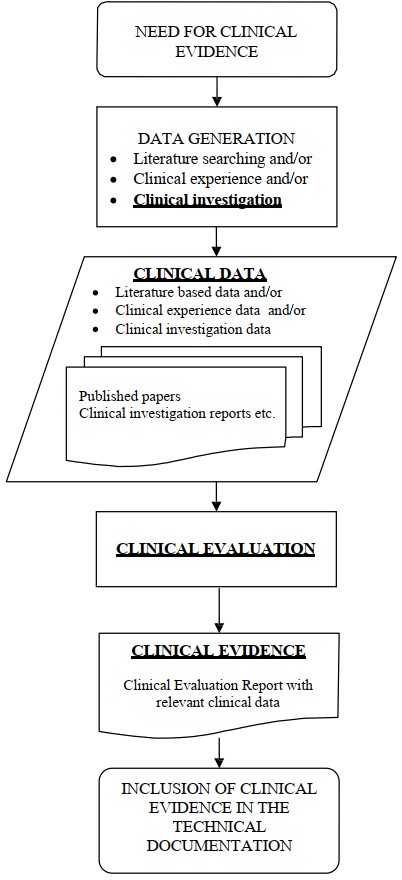
| CDE 的〈新版歐盟醫療器材法規 (MDR) 對於臨床評估之審查要求〉在第 2 頁開始,也有說明 Clinical data、Clinical evaluation 與 Clinical evidence 的定義與關係。 |
5. Clinical evaluation 的具體系統化程序?
本筆記首段提到,「臨床評估是一個事前規劃好的『系統化程序』」,接著第二、三段又說明「何時」與「為何要」做評估,但還是模模糊糊搞不清楚該如何下手?或者也懶得管那麼多背景資料,反正只要知道怎麼做就好?
好~接下來試著具體說明「臨床評估的系統化程序」。
臨床評估可以是一個反覆性的過程 (Iterative process)。
例如,可能初次評估不需要臨床試驗,但走一遍後發現,只有臨床試驗才可以取得必要的 Clinical data,這時就要修改計畫,再次執行。

圖/修改自 Sec. 6.3. of MEDDEV 2.7/1 (Rev. 4) 和 Figure 1 & Appendix C of IMDRF MDCE WG/N56FINAL:2019
上面流程圖 Stage 4 中的「EPs」全名為「Essential Principles of safety and performance of medical devices」,在不同時期、國家有不同的名稱和具體要求:
| 歐盟 | 美國 | 台灣 |
|---|---|---|
| MDR 時代的 EPs 稱為「General Safety and Performance Requirements (GSPRs)」,明訂於 MDR 的 Annex I。 | Substantial Equivalence (SE)。 相關筆記請參考〈如何使用 Multiple Predicates 來證明 Substantial Equivalence〉和〈如何用 Reference Devices 證明 510(k) 的實質相等性 (SE)〉。 | 產品實質等同性 (新申請產品及比較產品之異同處比對),和美國的 SE 相同。 |
總的來說,Clinical evaluation 目的是要透過臨床前 (Pre-clinical) 試驗、文獻探討,甚至臨床試驗,證明器材符合 EPs,或者可證明和類似品的相似度,最終達到法規、指引所定義的目的。
接下來,嘗試簡單筆記每個步驟的內容。
6. Stage 0 – 計畫 (Plan)
| 執行臨床評估的人員資格要特別注意! 例如,在 MEDDEV 2.7/1 (Rev. 4) 的「6.4. Who should perform the clinical evaluation? 」有特別規定,而 IMDRF MDCE WG/N56FINAL:2019 也說要由「Suitably qualified individual or individuals」執行,且須「Justify the choice of the evaluator(s) through reference to qualifications and documented experience」。 不過,目前好像沒有看到 MDR 相關規定。 |
| 除了本章節接下來的重點外,還可參考下列指引來建立臨床評估計畫: 1. IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) 的「Appendix G: A Possible Format for a Clinical Evaluation Report」; 2. MEDDEV 2.7/1 (Rev. 4) (Clinical Evaluation) 的「7. Definition of the scope of the clinical evaluation (Stage 0)」。 |
| 除了上述兩指引的參考內容外,MDR 在 Annex XIV Part A 的 Sec. 1(a) 也有規定 Clinical evaluation plan 最少要有的內容: 1. an identification of the general safety and performance requirements that require support from relevant clinical data; (識別需透過 Clinical data 支持的 GSPRs) 2. a specification of the intended purpose of the device; (預期用途的詳細說明) 3. a clear specification of intended target groups with clear indications and contra-indications; (明確說明預期使用族群,以及對應的適應症、禁忌症) 4. a detailed description of intended clinical benefits to patients with relevant and specified clinical outcome parameters; (詳細描述病患的預期臨床效益,以及相關的臨床結果參數) 5. a specification of methods to be used for examination of qualitative and quantitative aspects of clinical safety with clear reference to the determination of residual risks and side-effects; (詳細說明用於檢驗臨床安全性的定性與定量方法,以及殘餘風險、副作用的確定方法) 6. an indicative list and specification of parameters to be used to determine, based on the state of the art in medicine, the acceptability of the benefit-risk ratio for the various indications and for the intended purpose or purposes of the device; (一個規格清單,並基於當前醫學技術發展現況 (State of the art in medicine),來確定各種適應症與預期用途的利益風險比可接受度) 7. an indication how benefit-risk issues relating to specific components such as use of pharmaceutical, non-viable animal or human tissues, are to be addressed; and (說明如何解決特定方面的風險利益問題,例如,使用藥品、非活性動物或人體組織) 8. a clinical development plan indicating progression from exploratory investigations, such as first-in-man studies, feasibility and pilot studies, to confirmatory investigations, such as pivotal clinical investigations, and a PMCF as referred to in Part B of this Annex with an indication of milestones and a description of potential acceptance criteria. (一份臨床開發計畫,表明從探索性試驗到驗證性試驗的進展,以及符合附錄 XIV 第 B 部分所規定的 PMCF,此 PMCF 要列出里程碑與預計的允收標準。) – 關於上述第 8 點的「Clinical development plan」,依〈A Notified Body’s perspective on the clinical evaluation requirements under Regulation (EU) 2017/745 on the medical devices〉的 Figure 1 (Clinical process under the MDR) 說法,只有「all Class III and Class IIb devices intended to administer/remove medicinal substances (Article 61(2))」要遵守。 |
| 感謝神通廣大網友的回饋,我寫錯了「Clinical development plan (CDP) 的適用產品等級」!依照我引用的指引,是「所有等級 (All classifications)」醫療器材都要有 CDP,而非高風險的產品。 那 CDP 要寫啥內容?嗯…目前我只能簡單引用 Elsmar Cove 討論串「Clinical Development Plan – Advice Requested」中網友提到的做法, #7 PVC Barbie 網友說:「We have a CDP section in our CEP rather than a separate document. With that being said, it hasn’t been through MDR audit yet so…fingers crossed it will pass the test!」 #8 liz5572 網友跟著說:「Agreed that this doesn’t have to be a separate document. In the CEP, it can be as simple as a section titled “Clinical Development Plan” with a table with the name of the study (past and future/planned), purpose and status. It gives the reader a feel for what efforts have been taken, and what will be taken – and should align with the data presented in the CER and PMCF plan.」 |
在 Stage 0 時要先「畫靶」,例如:(節錄自 MEDDEV 2.7/1 (Rev. 4))
- 確認目標醫材的安全、臨床效能需求 (Define needs regarding clinical safety and clinical performance of the device);
- 若使用類似品,則評估是否有足夠的臨床資料證明與目標醫材的等同性 (In case of possible equivalence to an existing device, evaluate if there are clinical data available and determine equivalence);
- 透過差異分析,確認有哪些資料要透由臨床評估取得,以及確認是否有執行臨床試驗的必要 (Carry out a gap analysis and define which data still need to be generated with the device under evaluation, whether clinical investigations are necessary)。
此階段所訂出的「靶」,也就是「希望可以透過臨床評估釐清的問題」是後續步驟的基石。因此,至少要確認以下幾點。
| 我自己常覺得這部分很無聊,就只是用很多已知、且已出現在其他技術文件的資料來充版面。 但以制訂規定的專家學者角度來看,這部分其實是很重要的。因為如果在一開始時就可精準地界定出「靶」,可能就可避免耗時昂貴的臨床試驗了。關於此概念的說明,我引用澳洲 TGA 的指引如下: 「This will allow the clinical data requirements to be established more precisely in relation to the intended purpose of a device. Precision in this analysis and the choice of selected medical indications and target populations may reduce the amount of clinical data needed from additional clinical investigations.」 – Clinical evidence guidelines for medical devices (V3.0 November 2021) | Clinical investigations |
6.1 型號 / 設計特徵 / 目標族群 / 預期用途
這部分至少要釐清:
- 欲執行評估的型號;
- 有效能或安全疑慮的設計特徵 (Design features)。例如,是否含藥品、人類或動物成份等;還有
- 預期用途與應用 (Intended purpose and application)。例如,目標族群、適應症、警語、禁忌症、使用方法,或
- 其它製造廠所宣稱 (Claims)、且和安全、臨床效能、和/或功效有關的規格。
關於第 2、3、4 點,製造廠可由風險管理得到「Significance of any clinical risks that remain after design risk mitigation strategies have been employed」的資訊,並透過臨床評估去處理這類問題。
6.2 類似品 (Comparable devices)
用於臨床評估的相關資料可能來自於製造廠,也可能來自製造廠外的單位;可能是目標醫材本身 (Device in question) 的實驗/試驗/使用資料,但也可能來自製造廠宣稱的類似品 (Comparable devices,aka Equivalent device)。
如果不做臨床試驗的話,通常用於臨床評估的資料會來自製造廠外的類似品。若是這樣,製造廠就要先證明「類似品等同性」:
…it is demonstrated that the device subject to clinical evaluation for the intended purpose is equivalent to the device to which the data relate, in accordance with Section 3 of Annex XIV, and…
依據附錄 XIV 第 3 節,證明用於預期用途的臨床評估目標醫材,與臨床資料相關的醫材具等同性。
Medical Device Regulation (EU) 2017/745 | Sec. 3(a) of Article 61 (Clinical Evaluation)
一般會透過三方面來確認類似品的適用性:
- Clinical (Intended use):包含相關的臨床症狀 (Clinical condition)、疾病的嚴重程度和期數 (Stage)、用於病患身上 / 體內的位置 (Site of application to/in the body),還有病患與使用者族群等等;
- Technical:使用情境 (Condition of use)、設計規格、能量強度、佈建方式 (Deployment methods)、關鍵效能要求 (Critical performance requirements)、操作原理等等;
- Biological:生物相容性問題,例如,與病患組織、體液接觸的材料或物質、接觸時長 (Duration of contact)、降解產物 (Degration products) 與溶出物 (Leachables) 等等。
| 關於「類似品等同性」的證明, 1. 美國方面,可參考〈如何使用 Multiple Predicates 來證明 Substantial Equivalence〉和〈如何用 Reference Devices 證明 510(k) 的實質相等性 (SE)〉; 2. 台灣呢,其實跟美國幾乎一樣,可參考竹科管局〈請問在台灣查驗登記送審時,實質等同性的判定應檢附哪些資料〉和 TFDA〈醫療器材類似品判定流程及函詢申請說明〉; 3. 歐盟 MDCG 有針對此問題推出指引〈Guidance on clinical evaluation–Equivalence〉(MDCG 2020-5,2020); 4. IMDRF 的〈Clinical Evaluation〉(WG/N56FINAL:2019) 在附錄 A 也有依等同性三方面 (Intended use / Technica l /Biological) 列出評估項目; 5. MDR 在 Annex XIV 的 Sec. 3 of Part A (Clinical Evaluation) 有規定證明的要點; 6. 澳洲 TGA〈Clinical evidence guidelines for medical devices〉(V3.0 Nov 2021) 在「Part 2 – Special topics」(P.47) 有討論。 關於這個,我們之後會專門整理一篇來分享。 |
6.3 相關領域的 State of the art
要確認相關領域的 Current knowledge 或 State of the art。例如,現行的標準、指引、實際醫療情境下相關儀器的運作方式、Benchmark devices、醫療替代方案等。
什麼是 State of the art (SOTA)?
IMDRF/GRRP WG/N47 provides the following definition:
Developed stage of current technical capability and/or accepted clinical practice in regard to products, processes and patient management, based on the relevant consolidated findings of science, technology and experience.
Note: The state-of-the-art embodies what is currently and generally accepted asgood practice in technology and medicine. The state-of-the-art does not necessarily imply the most technologically advanced solution. The state-of-the-art described here is sometimes referred to as the “generally acknowledged state-of-the-art”.
Guidance on sufficient clinical evidence for legacy devices (MECG 2020-6) | Sec. 1.2. Additional terms not defined in MDR Article 2
要從哪取得證明 SOTA 的資料呢?可參考以下來源:
– European medical societies/national medical organizations (e.g. European Society of Cardiology);
– literature searches (most current/recent publications relevant to indications);
– international guidance (relevant to the EU population) to national guidance documents (e.g. guidance from the UK National Institute for Clinical Excellence);
– Real-World Evidence (e.g. registry data);
– non-inferior model (statistical) analysis of Post-Market Surveillance (PMS) data;
– physician surveys/usage data.
A Notified Body’s perspective on the clinical evaluation requirements under Regulation (EU) 2017/745 on the medical devices | Page 36
6.4 資料來源與資料型態
依照器械的設計、預期用途與風險等級,會有不同的需求。
例如,若產品使用現有的成熟技術 (Well-established technology),則可能用相關標準、文獻探討 (Literature review)、類似品的臨床使用經驗 (Clinical experience of comparable devices) 即可。但若是高風險器械,或創新的產品,則很可能要有臨床試驗資料 (Clinical investigation data) 才足夠。
建立好計畫,並確認臨床評估範圍 (Scope) 後,就可依計畫執行臨床評估。
以下是臨床評估正式實施的三階段。
7. Stage 1 – 資料識別 (Identification)
一般有三個來源可用於此階段來取得相關資料:
- Literature searching (文獻搜尋)、
- Clinical experience (臨床使用經驗,aka Real World Data),和
- Clinical investigation (臨床試驗),
以下將簡單說明此三來源。
7.1 Literature searching
此方法可用來搜尋已發表、但製造廠尚未掌握的 Clinical data (臨床資料),這些資料可能
- 直接和目標醫材本身相關 (Relate directly to the device in question),例如,製造廠外的其他單位拿目標醫材去執行的臨床試驗,或目標醫材的不良事件報告;
- 也可能和類似品 (Comparable device) 相關。
在執行 Literature searching 前,須先建立 Literature search protocol,內容包含:
- Source:除說明資料來源外,還須解釋為何選擇這些來源;
- Extent:資料庫的搜索策略,會影響在特定資料來源內搜尋到多少文獻;
- Selection criteria:資料的納入/排除準則,並且須說明準則的設立原因;
- Duplicated data addressing strategies:處理不同文獻中重覆出現相同資料的策略。
至於,文獻搜尋有哪些來源呢?以下是實證醫學常用的分類:
| 證據力等級 | 5S Model (Haynes RB 於 2006 年提出) | 階層說明 | 資料庫 |
|---|---|---|---|
| 5 (證據力最強) | Systems (系統) (Computerized decision support) | 針對某些已有標準療法的疾病,醫師可直接參考此電腦自動化的決策系統提供的治療方案。 | – |
| 4 (證據力次之) | Summaries (總結) (Critically-appraised topics, Evidence-based guidelines) | 由各領域的專家學者整理單一主題的多篇研究或系統回顧而成,有很具體的研究成果與臨床建議,同時也有同儕審查機制,因此正確性高,且具權威性。 | DynaMed UpToDate BMJ Clinical Evidence |
| 3 | Synopses (精要) (Critically-appraised journal articles) | 相較於 Summaries 是整理多篇研究或系統回顧而成,Synopses 只針對已有共識的單一文章,將其整理成容易閱讀的一至兩頁摘要內容。 | Database of Abstracts of Reviews of Effects Evidence-Based Medicine Evidence-Based Nursing ACP Journal Club |
| 2 | Syntheses (統整) (Systematic reviews) | 就特定問題的系統性評論文獻,通常可由此階層的文獻得知某一主題的最新發展,故是 5S 中最為重要的。 | Cochrane Database of Systematic Reviews BMJupdates+ PubMed Clinical Queries |
| 1 (證據力最弱) | Studies (原始研究文獻) (Original journal articles) | 依 Oxford Centre for Evidence Based Medicine 將證據強度 (OCEBM Levels of Evidence) 由高到低排序:RCTs > Cohort studies > Case-control studies > Case series | PubMed CINAHL PsycInfo |
在查找資料時,通常由證據力高的來源開始找。
若只剩「Studies」等級的資料可用,通常代表這方面的問題研究還不夠多,可能是因為大家尚未關注此議題,或正反意見都有還未形成共識。
Literature search protocol 的執行結果必須整理成 Literature search report,而 Literature search protocol 要附在報告中,若有任何偏離於 Protocol 的情況,也要在報告中闡明。
至於 Literature search report 內容要多詳細?IMDRF 指引說:
It is important that the literature search is documented to such a degree that the methods can be appraised critically, the results can be verified, and the search reproduced if necessary.
要詳細到
1) 文獻搜尋方式要足以被嚴謹地評讀,
2) 結果可被驗證,以及
3) 必要時,可以再現搜尋的程度。
Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019) | 6.1 Data generated through literature searching
| Literature search report 的參考格式: 1.〈Clinical Evaluation〉(IMDRF MDCE WG/N56FINAL:2019) 的「Appendix B: A Possible Format for the Literature Search Report」; 2.〈Clinical Evaluation〉(MEDDEV 2.7/1,Rev. 4) 的「A5. Literature search and literature review protocol, key elements」。 |
因此,搜尋完文獻,至少會有三份文件:
- Literature search protocol;
- Literature search report;
- 與目標醫材或類似品相關的文獻完整副本 (不可只放文獻摘要,因摘要可能缺乏足夠的細節以執行接下來的評讀、分析與總結) (Sec. 8.2. of MEDDEV 2.7/1 (Rev. 4))。
7.2 Clinical experience
| Clinical experience (臨床使用經驗) 的定義已於前面筆記討論過了。還是要特別提醒一下,在 MDR 時代,Clinical experience 的相關文獻須「經同儕審查」才可用於臨床評估。 |
相較於 Clinical investigation 會有納入/排除準則來篩選受試者,以提升受試族群的同質性 (Homogenous)、減少變因,進而得到較可信 (Confidence) 的結論;Clinical experience 可提供更為複雜、異質性 (Heterogeneous) 更高的族群使用經驗,可稱為「Real world experience」。
透過 Clinical experience 的來源,可取得以下幾個利於臨床評估的資料:
- 較不常見,但嚴重的不良事件資訊;
- 關於安全、臨床效能、和/或功效資訊的長期資訊;
- End user (終端用戶) 的 Learning curve (學習曲線)。
| 以下是常用的 Adverse event report databases: USFDA: Manufacturer and User Facility Device Experience (MAUDE)、Total Product Life Cycle (TPLC) TGA Australia: Database of Adverse Event Notifications (DAEN);Recall actions database Germany 聯邦藥品與醫療器械研究所 (Bundesinstitut für Arzneimittel und Medizinprodukte,BfArM) Japan PMDA: 不具合が疑われる症例報告に関する情報 |
Clinical experience 對於有歷史 (Long standing)、低風險且基於成熟技術 (Well-established technology) 的產品來說更為重要,因為不太會有科學文獻或臨床試驗以此類產品作為研究主題。(Sec. 6.2 of IMDRF MDCE WG/N56FINAL:2019)
| MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) 將上述由 Risk management activities 和 PMS programmes 得到的資訊,以及上市前的臨床試驗,歸類在「Data generated and held by the manufacturer」;而把 Literature searching 得到的資訊歸類於「Data not held by the manufacturer」。 更多細節可看 MEDDEV 2.7/1 第 8.1 及 8.2 章節。 |
而 Registry (註冊登記 / 登記,aka. Registry study (註冊登記研究 / 註冊研究 / 登記研究)) 是 Clinical experience 一個有用的資訊來源。
什麼是 Registry 呢?
A clinical registry is a computer database that collects information about your health and the care you receive as a patient. The data in the registry comes from the information your healthcare provider collects while providing your care and is added to information on other patients who are similar to you.
Registries collect information on large numbers of similar patients. This information may include things like: a patient’s reasons for seeking care, treatments they received, and how well they did over time.
OrthoInfo | What Is a Clinical Data Registry?
Device registry: an organised system that uses observational study methods to collect defined clinical data under normal conditions of use relating to one or more devices to evaluate specified outcomes for a population defined by a particular disease, condition, or exposure and that serves predetermined scientific, clinical or policy purpose(s).
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 4. Definitions
Registry 可以做什麼?
… your doctor can help you choose treatments that have tended to work well for patients like you. If you have an implant, for example, information from registries may also be used to identify poorly performing implants and can alert your hospital about recalls to those components.
OrthoInfo | What Is a Clinical Data Registry?
… Medical device manufacturers and pharmaceutical developers use registries to track and understand the effectiveness, safety, and value of medical devices or therapies and drugs entering or on the market.
ArborMetrix | The Basics of Clinical Data Registries
Registry 有哪些種類?
There are many types of registries. Some track patients who have a particular disease or condition. Others track the performance of medical devices such as artificial joints.
OrthoInfo | What Is a Clinical Data Registry?
| 關於「Registry」有以下三本指引可參考: 1. 〈Principles of International System of Registries Linked to Other Data Sources and Tools〉(IMDRF/REGISTRY WG/N33 FINAL: 2016) 2. 〈Methodological Principles in the Use of International Medical Device Registry Data〉(IMDRF/Registry WG/N42FINAL:2017) 3. 〈Tools for Assessing the Usability of Registries in Support of Regulatory Decision-Making〉(IMDRF/Registry WG/N46 FINAL: 2018) (可能~) 我們之後會專門整理一篇來分享。 |
| TGA 指引 提供兩個 Australian device registries:Australian National Orthopaedic Association National Joint Replacement Registry (AOANJRR),以及 Victorian Cardiac Outcomes Registry。 |
〈Clinical evaluation〉(IMDRF MDCE WG/N56FINAL:2019) 也在第 6.2 章節 (Data generated through clinical experience) 提到,「Post market surveillance reports」是一個可能的資料來源,因為報告中會有:
- 法規現況 (Regulatory status):在哪個國家上市,何時開始供貨;
- 法規活動 (Regulatory actions):有無召回 (Recalls) 、通知 (Notifications)、現場安全矯正措施 (Field Safety Corrective Actions, FSCA)、Vigilance reports;
- 不良事件列表 (Tabulation of adverse events)、發生率,和 Trend reports。
但要特別注意所搜尋到的不良事件是否可能在目標醫材上發生,還有若是這些不良事件沒有在現有的風險管理中被考慮到,則要控管此風險 (例如,變更設計、改標識),並且文件化。
7.3 Clinical investigation
有超多要考慮的,本筆記暫且跳過不寫了…(沒力了 orz)
順帶一提,依照 MDR 的 Article 61 Sec. 4 的規定,Implantable devices 和 Class III devices 都必須執行臨床試驗,除非符合一些條件。
想知道怎樣才不用做臨床試驗嗎?請參考另一篇筆記〈我的醫材需要臨床試驗 (Clinical investigation) 嗎〉。
8. Stage 2 – 資料評讀 (Appraisal)
8.1 Appraisal 目的
The purpose of undertaking appraisal of the data is to understand the merits and limitations of the clinical data. Each piece of data is appraised to determine its suitability to address questions about the medical device, and its contribution to demonstrating the safety, clinical performance and/or effectiveness of the device (including any specific claims about safety, clinical performance and/or effectiveness).
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | 7.0 Appraisal of clinical data (Stage 2)
因此,Stage 2 目的是,評讀每個在 Stage 1 (Identification) 找到資料的:
- Suitability (適用性):是否適用於解決於 Stage 0 找出要透過臨床評估解決的問題;以及
- Contribution (貢獻度):證明醫材安全、臨床效能、和 / 或功效的能力。
8.2 The appraisal plan
To ensure systematic and unbiased appraisal of the data, the evaluators should set up an appraisal plan that describes the procedure and the criteria to be used for the appraisal.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 9.2. The appraisal plan
評估者 (Evaluator) 須在進行 Stage 2 前先建立資料評讀原則 (Appraising criteria),接著才依此原則開始執行評讀。
一開始,要先檢查 Stage 1 所用的資料識別「方法」是沒問題的,接著再評估 Clinical data 內與安全或效能相關的結果,有多大程度是和醫材有關。或者,可能有些結果其實和醫材沒關係,而是被其它因素所影響,例如:
- Confounding influences (E.g., Natural course of the underlying medical condition、Regression to the mean、Concomitant treatment(s))、
- Bias、
- Random error、
- Inadequate disclosure of information、
- Misinterpretation。
但 MEDDEV 2.7/1 (Rev. 4,Sec. 9.3.1.) 說:
Some papers considered unsuitable for demonstration of adequate performance because of poor elements of the study design or inadequate analysis may still contain data suitable for safety analysis or vice versa.
也就是說,可能某 Clinical data 對證明產品功效來說,不是一個好的佐證資料,但卻可用來證明安全性,反之亦然。
MEDDEV 2.7/1 (Rev. 4) 在 9.1 章節 (General considerations) 說明,Stage 1 找到的文獻,其不確定性 (Uncertainty) 會被下面兩方面影響:
- Methodological quality:要確認每篇文獻的方法,這會影響資料的科學效度 (Scientific validity);和
- Relevance:要確認資料與待解決問題的相關性。
如前所說,此階段的「Uncertainty」會被「Scientific validity」和「Relevance」影響,因此 Appraisal plan 要涵蓋這兩點。
另外,還要說明如何評估資料的「貢獻權重」(Contribution)。
那有沒有什麼方法可以來評估「Scientific validity」、「Relevance」、「Contribution」呢?
目前沒有一定的評讀方式,但可參考以下指引:
- IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) 的「Appendix E: Some Examples to Assist with the Formulation of Criteria 」、「Appendix F: A Possible Method of Appraisal」;以及
- MEDDEV 2.7/1 (Rev. 4,Clinical Evaluation) 的「9.3. Conduct of the appraisal」。
雖說沒有一定的評讀方式,但上兩份指引還是有建議的 Appraisal 重點。
8.3 Appraisal 重點
8.3.1 Scientific validity (科學效度) 評估重點
- 上市前/後的臨床試驗:例如,要看樣本數 (Sample size)、檢定力 (Power)、評估指標 (endpoints)、納入/排除準則 (Inclusion and exclusion criteria)…等 (Sec. 9.3.1.a. & 9.3.1.b. of MEDDEV 2.7/1 (Rev. 4));
| TGA 指引 提到 (P.28): 1. Single arm studies (and other study designs) with no comparator arm are generally considered inadequate evidence. 2. Comparisons of datasets obtained through different methodologies (for example, a case series using the subject device with standard of care outcomes established from a literature search) are generally considered poor quality evidence and may be subject to greater scrutiny, as necessary, when assessing whether that data supports compliance with the EPs. 3. Clinical safety and performance should generally be expressed in terms of person-centred outcomes, such as mortality, morbidity, adverse events, and patient reported outcome measures (PROMs). 4. Where study findings are expressed in terms of markers or intermediate measures of safety and performance, a clinically reasoned argument should be provided linking the study findings with patient centred outcomes. |
- Vigilance data, device registry data, case series, patient dossiers, and other use data:例如,當使用 Vigilance data 時要注意有無「低報/少報 (Under-reporting or lack of reporting)」不良反應或併發症;而當使用此類的其它資訊來源時,要注意更多問題,像是「Have all the patients been considered?」或「Are the patients representative of the use of the device?」等等… (Sec. 9.3.1.c. of MEDDEV 2.7/1 (Rev. 4));
- Data processing and statistics data:例如,資料轉換方式是否適當?排除的資料是否合理?…等等 (Sec. 9.3.1.d. of MEDDEV 2.7/1 (Rev. 4));
- Quality assurance:是否遵守 GCP (E.g., EN ISO 14155) …等 (Sec. 9.3.1.e. of MEDDEV 2.7/1 (Rev. 4));
- Report quality:要評估所收集到的資料中,是否有充分描述使用的方法、是否充分揭露相關數據、結論是否與結果一致…等等 (Sec. 9.3.1.f. of MEDDEV 2.7/1 (Rev. 4))。
| Sec. 9.3.1.f. of MEDDEV 2.7/1 (Rev. 4) 提到: 「Possible conflicts of interest of the authors of the publications should also be taken into consideration.」 這在 TGA 指引 也有說 (P.29): 「Studies conducted by manufacturers or sponsors, or those who have received funding or support from manufacturers or sponsors, will be considered on their merits. Peer reviewed articles should clearly identify any conflicts of interest (actual or perceived). It is accepted that certain studies require support from manufacturers (such as large-scale pre-market approval studies) or will be conducted by manufacturers (such as PMCF studies). A discussion of the extent of involvement of manufacturers or sponsors should form part of the study report and the critical analysis contained in the CER.」 |
8.3.2 Relevance 評估重點
要考慮所找到的資訊,對於證明 Safety、Clinical performance、Effectiveness 是直接或間接的,還要考慮 Benefit/risk profile 是否符合 Current knowledge/State of the art,以及是否有發現新的危害 (Hazards)…等等。
MEDDEV 2.7/1 (Rev. 4) 在 9.3.2.c. 章節 (How to determine the relevance of a data set for the clinical evaluation) 列出下面四大項評估重點,每一大項又細分成數個小項:
- To what extent are the data generated representative of the device under evaluation? (是用目標醫材進行實驗所得到的資料?還是用類似品?還是…)
- What aspects are covered? (這些資料可用來證明效能?安全?或證明製造廠的宣稱 (Claims)?還是…)
- Are the data relevant to the intended purpose of the device or to claims about the device? (這些資料可代表完整的預期用途 (Entire intended purpose)?或只涵蓋部分用途,甚至和目標醫材的預期用途一點關係都沒有?)
- If the data are relevant to specific aspects of the intended purpose or claims, are they relevant to a specific model, size, or setting of the device? user group? medical indication (if applicable)? age group? gender? type and severity of the medical condition? range of time? (承第 3 題,若只和部分預期用途、製造廠宣稱相關,那就要釐清是和哪部分相關。)
8.3.3 Contribution 評估重點
評估完「Scientific validity」和「Relevance」後,要開始評估資料的「貢獻權重」。
因醫療器材種類態樣繁雜,MEDDEV 2.7/1 (Rev. 4) 在 9.3.3 章節 (How to weight the contribution of each data set) 時說:
There is no single, well established method for weighting clinical data.
只有提到,一般來說下面這類 Clinical data 貢獻度是最高的:
…clinical data should receive the highest weighting, when
1) generated through a well designed and monitored randomized controlled clinical investigation (also called randomised controlled trial),
2) conducted with the device under evaluation in its intended purpose, 3) with patients and users that are representative of the target population.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 9.3.3. How to weight the contribution of each data set
8.3.4 其它評估重點
注意 Clinical data 是否符合相關法規或標準 (例如,ISO 14155),以及是否適用於正在申請上市的使用族群。
因為所收集到的 Clinical data 可能來自不同國家,因此除了上面提到要注意是否符合相關法規或標準外,還要考慮內在 (Intrinsic)/外在 (Extrinsic) 因子:
1. Intrinsic factors: human genetic characteristics or demographic factors, such as race, age, gender, etc.
2. Extrinsic factors: clinical practice, social environment, natural environment, cultural factors, life behavioral factors, rare or regional diseases, etc.
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | Appendix D: Considerations for the Application of Clinical Data Generated from Different Jurisdiction(s)
8.4 Appraisal 文件化程度
既然訂好 Appraisal plan,就要嚴格遵守,並且要針對全文 (Full text) 去評讀,而非只看摘要或總結 (Not abstracts or summaries)。
也就是說要評讀:
…all of the contents,
the methodology employed,
the reporting of results,
the validity of conclusions drawn from the investigation or report, and
evaluate any limitations and potential sources of error in the data.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 9.3. Conduct of the appraisal
上面這些活動都要文件化的內容,必須足以讓其它人可以嚴謹地審查 (Critically reviewed by others)。
9. Stage 3 – 資料分析 (Analysis)
9.1 Analysis 目標
此階段的目標是:
… to make a benefit/risk determination if the appraised data sets available for a medical device collectively demonstrate the safety, clinical performance and/or effectiveness of the device in relation to its intended use.
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | 8.0 Analysis of the clinical data (Stage 3)
The goal of the analysis stage is to determine if the appraised data sets available for a medical device collectively demonstrate compliance with each of the Essential Requirements pertaining to the clinical performance and clinical safety of the device, when the device is used according to its intended purpose.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 10.1. General considerations
若經由前兩階段 (Identification & Appraisal) 篩選出的資料,在此階段都一致顯示相同的結果,則關於安全、臨床效能、和/或功效的確信程度 (Certainty) 就會提高;反之,若發現有不同的結果,就要去分析了解原因 (例如,可能某些在 Stage 2 評讀較低的資料才會顯示出這些不同的結果)。
| 有關 Risk/benefit profile、Benefit-risk ratio、Benefit/risk determination 等等的利益與風險評估,請參考另篇筆記「醫療器材上市前審查考量之利益與風險權衡要素 (Benefit-Risk evaluation)」。 |
9.2 Analysis 重點
評估者 (Evaluator) 要考量資料的以下重點:
- 可用定性 (Qualitative) 或定量 (Quantitative) 的 Weighting criteria,若採用定性方法,須說明理由 (Section 10.2.a. of MEDDEV 2.7/1 (Rev. 4));
- 接觸此醫材的病患數量與特徵 (Number and characteristic of patients exposed to the medical device);
- 監控的形式與是否充分 (Type and adequacy of patient monitoring);
- 不良事件的數量與嚴重性 (Number and severity of adverse events);
- 對已識別危害的相關風險的估計是否充分 (Adequacy of the estimation of associated risk for each identified hazard);
- 被診斷/治療的疾病的嚴重性或自然發展歷史 (Severity and natural history of the condition being diagnosed or treated);
- 是否有替代的診斷方法/治療方法/護理標準 (Availability of alternative diagnostic modalities or treatments and current standard of care);
- 醫材使用者/接受者的後續追蹤持續時間 (Duration of follow-up of device recipients)。
9.3 Analysis 結論
由前面兩階段,將收集到的 Clinical data (臨床資料) 透過 Clinical evaluation (臨床評估) 整理成 Clinical evidance (臨床證據) 後,製造廠要下結論確認 Clinical evidance 是否足以支持產品符合 EPs 的要求。
若覺得證據不足,就可能要想辦法取得更多資料,例如,進行臨床試驗,或者擴大文獻搜尋的範圍 (Broaden the scope of literature searching)。
因此,臨床評估可能是一個反覆性的過程 (Iterative process)。
最終,評估者要做出以下結論:
– the medical device performs as intended by the manufacturer;
– the medical device does not pose any undue safety concerns (undesirable side-effects) to either the recipient or end-user;
– any risks associated with the use of the device are acceptable when weighed against the benefits to the patient (benefit/risk profile);
– the clinical evidence demonstrates conformity with relevant Essential Principles; and
– whether post market clinical follow up or post approval study is necessary;
是否有殘餘風險或未確定、未回答的問題 (例如,Rare complications, uncertainties regarding long-term performance, safety under wide-spread use) 要透過 Post-market surveillance (上市後監督,PMS) 來解決
– whether the safety characteristics and intended purpose of the medical device requires training of the end-user.
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) | 8.0 Analysis of the clinical data (Stage 3) & 6.2 & 7 of Appendix G
還要:
Determine if additional clinical investigations are necessary.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 10.2. Specific considerations
最後,要確認 Clinical evaluation / Information materials supplied by the manufacturer / Risk management documentation 三者的一致性。
如果 Clinical data 不足以用來證明產品符合 Essential Principles (或 GSPR、SE),製造廠還有機會透過 Performance evaluation、Bench testing、Pre-clinical evaluation 作為佐證的方式,以風險管理報告為基礎,針對下列三重點提出有力的 Justifications 來說明產品是符合 Essential Principles (或 GSPR、SE) 的:(Sec. 10.3. of MEDDEV 2.7/1 (Rev. 4))
- The device/body interaction,
- The clinical performances intended and
- The claims of the manufacturer have to be specifically considered.
10. 上市後更新時注意事項
更新時,要特別注意:
If the benefit/risk profile, undesirable side-effects (whether previously known or newly emerged) and risk mitigation measures are still
– compatible with a high level of protection of health and safety and acceptable according to current knowledge / the state of the art;
– correctly addressed in the information materials supplied by the manufacturer of the device;
– correctly addressed by the manufacturer’s current PMS plan;
If existing claims are still justified;
If new claims the manufacturer intends to use are justified.
MEDDEV 2.7/1 (Clinical Evaluation,Rev. 4) | 6.2.3.b. General considerations on updating the clinical evaluation
若上面的答案是「否」,就要採取必要的 Corrective and preventive action。
| 還可參考 MEDDEV 2.7/1 (Rev. 4) (Clinical Evaluation) 的「7. Definition of the scope of the clinical evaluation (Stage 0)」,例如考慮: 1. Whether the claim of equivalence to an existing device is still appropriate; 2. Whether new clinical data is still available for the device under evaluation, and for the equivalent device (if equivalence is claimed), …等等。 |
11. CER 其它注意事項
11.1 CER 完整性
IMDRF MDCE WG/N56FINAL:2019 (Section 9,The Clinical Evaluation Report) 與 MEDDEV 2.7/1 (Rev. 4) (Section 11,The clinical evaluation report) 中提到,Clinical Evaluation Report (CER) 應詳細到可讓獨立單位 (Independent party, e.g. regulatory authority or notified body) 將此視為一個獨立文件 (Standalone document),以了解:
- Search criteria adopted by the evaluators,
- Data that are available,
- All assumptions made and
- All conclusions reached.
因此,CER 至少要包含以下資訊:
- the technology on which the medical device is based, the intended use of the medical device and any claims made about the device’s safety, clinical performance and/or effectiveness;
| 雖說要「視為獨立文件」,但也不用包山包海地再寫一遍所有細節,IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) 在 Appendix G (A Possible Format for a Clinical Evaluation Report) 寫到: 「Provide a concise physical description of the medical device, cross referencing to relevant sections of the manufacturer’s technical information as appropriate.」 因此,仍是可以引用其它技術文件啦。 |
- the nature and extent of the clinical data that has been evaluated; and
- how the referenced information (recognised standards and/or clinical data) demonstrate the safety, clinical performance and/or effectiveness of the device in question.
TGA 指引引用兩本〈Clinical Evaluation〉— IMDRF MDCE WG/N56FINAL:2019 的「Appendix G: A Possible Format for a Clinical Evaluation Report」以及 MEDDEV 2.7/1 rev. 4 的「A9. Clinical evaluation report」,建議 CER 可以有以下內容:(細節請見上述兩本指引)
- General details
- Description of the medical device and its intended application
- Intended therapeutic and/or diagnostic indications and claims
- Context of the evaluation and choice of clinical data types
- Summary of relevant pre-clinical data
- Discussion regarding comparable devices including substantially equivalent devices
- Summary of the clinical data and appraisal
- Data analysis
- Conclusions
- Name, signature and curriculum vitae of the clinical expert and date of report
大家可以整理成如下查檢表:
| Section | Included (Yes、No、N/A) | Author(s) | Page(s) |
|---|---|---|---|
| 1. General details and device description -including GMDN, UDI, lineage and version (if applicable) | |||
| 2. Intended purpose, indications and claims | |||
| 3. Developmental context and state of the art | |||
| 4. Regulatory status in other countries (including evidence and supporting documents) | |||
| 5. Summary of relevant pre-clinical data (if applicable) | |||
| 6. Demonstration of substantial equivalence or comparability (if applicable) | |||
| 7. Summary and appraisal of clinical data | |||
| 8. Data analysis and benefit-risk analysis | |||
| 9. Conclusions | |||
| 10. Name, signature and curriculum vitae of clinical expert and date of report | |||
| 11. Risk assessment and management documents | |||
| 12. IFU, labelling and other documents supplied with the device | |||
| 13. Full clinical investigation reports | |||
| 14. Literature search and selection strategy | |||
| 15. Full text of pivotal articles from the literature review | |||
| 16. Post-market surveillance reports | |||
| 17. Additional relevant information on the device (if applicable) |
10.2 Evaluator 審核與資格
CER 要讓評估者 (Evaluator) 簽名並壓日期,並附上選擇此評估者的理由。
| IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) 在 Appendix G (A Possible Format for a Clinical Evaluation Report) 提供 CER 的模板供大家參考。 |
10.3 Clinical data 分類與列表
IMDRF MDCE WG/N56FINAL:2019 (Clinical Evaluation) 在第 5 章節 (Summary of the clinical data and appraisal) 建議,將所使用的 Clinical data 依其對應所解決的問題類別,分為 Safety、Clinical performance、Effectiveness (安全、臨床效能、和 / 或功效) 三類。
將 Clinical data 分為三類後,可再將其依照 Suitability 和 Contribution 由高到低排序。
10.4 常見錯誤
TGA 指引 (P.41) 提供了 CER 常見的缺失:
- 缺少 CER 必須要素和/或附件 (Absence of the required components of the CER and/or referenced attachments and appendices missing) (請搭配〈Clinical Evaluation 101:關於臨床評估的基本了解〉檢查是否 CER 已包含所有必要內容)
- 不同文件間的預期用途、適應症、宣稱不一致 (Intended purpose(s), indication and claims inconsistent between documents – for example, the application, IFU and CER list different intended purpose(s))
- Clinical data 不支持預期目的、適應症、宣稱 (Intended purpose(s), indication and claims not supported by clinical data)
- 缺少各國的法規歷史資訊 (Lack of information about the regulatory history of the device in other countries, for example recalls, withdrawals, removals from market, suspensions and cancellations and the reasons for these in any jurisdiction)
- (若適用) 缺少類似品資訊,或不足以證明兩者的實質相等性 (Information on comparable devices not included and/or substantial equivalence not demonstrated (if relevant))
- (若適用) 目標產品或類似品的臨床試驗、文獻或上市後資料不足或不完整 (Insufficient or incomplete clinical investigation(s) data, literature and post-market data with the device or comparable device (if relevant))
CER「應詳細到可讓獨立單位 (Independent party, e.g. regulatory authority or notified body) 將此視為一個獨立文件 (Standalone document)」,要記錄下所有過程,不是自己做完就算了。因此,CER 若缺少以下文獻探討 (Literature review) 相關項目,也會被視為缺失:
- 沒有文件化的方法,和/或沒有呈現出完整的文獻探討過程 (No documented method and/or no demonstrated comprehensive literature review)
- 文獻搜尋的相關資訊不足,或文獻搜尋方式品質不良導致無法再現/了解文獻探討的策略 (Insufficient information and/or poor-quality search protocol that result in inability to reproduce or understand the literature review strategy )
- 未解釋為何引用文獻的相關性 (Provision of a multitude of publications with little or no explanation as to why they are of relevance)
- 沒有定義清楚所用的設備或使用方式 (No identification of device used or indication for use in articles reviewed)
- 沒總結每一個納入文獻的研究特點 (可用表格總結各篇文獻的重要研究結果,像是這樣) (No summary of study characteristics and findings for each included article)
很少,甚至沒有對臨床試驗資料、文獻探討、上市後資料進行綜合 (Synthesis) 且嚴謹的 (Critical) 評估,也是一類缺失。例如:
- 沒有關於「強度」的討論 (No discussion of relative strengths of the data, for example randomised controlled trials, case control studies, case series)
- 證明實質等同性的資料不足 (Substantial equivalence covering technical characteristics, biological characteristics and clinical use not established to validate the data for a different device (i.e. comparable device) to the device under review)
- 缺少關於「效度」的討論 (Lack of discussion of the validity or otherwise of outcome measures used)
- 沒有臨床專家認可差異不會對安全與功效產生不良影響 (No endorsement by the clinical expert that the differences will not adversely affect the safety or performance of the device)
最後,還有以下常見的缺失:
- 對整體證據的評論與總結不足 (Inadequate critique and summary of the totality of evidence provided for the device)
- 沒有上市後資料 (No post-market data including adverse events, vigilance reports, complaints, failures in cases where this information is available)
- CER 未被臨床專家簽屬核准,且/或沒有日期或過期 (CER not endorsed/signed by clinical expert and/or CER not dated or out-dated)
- 臨床專家不適合 (Inappropriate selection of clinical expert/s)
- 沒提供臨床專家的履歷 (CV of clinical expert/s not provided.)
4 留言
您好,謝謝您如此詳細的說明,獲益良多
但想請問您如果是現行台灣法規,且產品為一類低風險的產品
此部分應該如何撰寫,才能提供完整性的臨床評估報告呢?謝謝您~
嗨 Jill,
很抱歉,我沒處理過台灣第一等級的醫材,無法給什麼明確的建議。
不過,依照〈第一等級醫療器材查驗登記申請文件檢送簡表〉,似乎為了符合第七項要求「臨床前測試之檢驗規格與方法及檢驗成績書」,有機會需要臨床評估。
但…我猜想需要臨床評估的機會不大,通常是照著一些產品性能標準進行測試即可。
因為,就算是第二、三等級的醫材,通常也是在「沒有類似品」的情況下,才會要求附上「臨床評估報告」。(請參考〈第二、三等級國產(輸入)醫療器材查驗登記形式審查查檢表〉第十八項第 2 條要求)
實在很佩服您的筆記,太厲害了!
下面這個內容,我想可以再確認一下:
“關於上述第 8 點的「Clinical development plan」,依〈A Notified Body’s perspective on the clinical evaluation requirements under Regulation (EU) 2017/745 on the medical devices〉的 Figure 1 (Clinical process under the MDR) 說法,只有「all Class III and Class IIb devices intended to administer/remove medicinal substances (Article 61(2))」要遵守。”
請看看所引用文件〈A Notified Body’s perspective on the clinical evaluation requirements under Regulation (EU) 2017/745 on the medical devices〉的 Table 1. “Clinical development plan” 應該仍是所有等級都需要有的,但”linical development strategy”才僅要求「all Class III and Class IIb devices intended to administer/remove medicinal substances (Article 61(2))」要遵守。” Figure 1. 很容易引起誤解。淺見,供參考
感謝分享。