筆記目錄
1. 前言
上一篇筆記〈Clinical Evaluation 101:關於臨床評估的基本了解〉主要內容來自下列歐系指引:(對,我也把 IMDRF 當成歐洲 XD)
- Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019)
- Clinical Evaluation (MEDDEV 2.7/1,Rev. 4)
- Medical Device Regulation (EU) 2017/745
而本篇筆記將以澳洲 TGA〈Clinical evidence guidelines for medical devices〉(V3.0 Nov 2021,下稱「TGA 指引」) 為主,補充之前沒提到的 Clinical Evaluation 資訊 (跳過 TGA 指引中澳洲的法規要求),尤其特別著重在「資料識別 (Identification) /評讀 (Appraisal) 與分析 (Analysis)」。
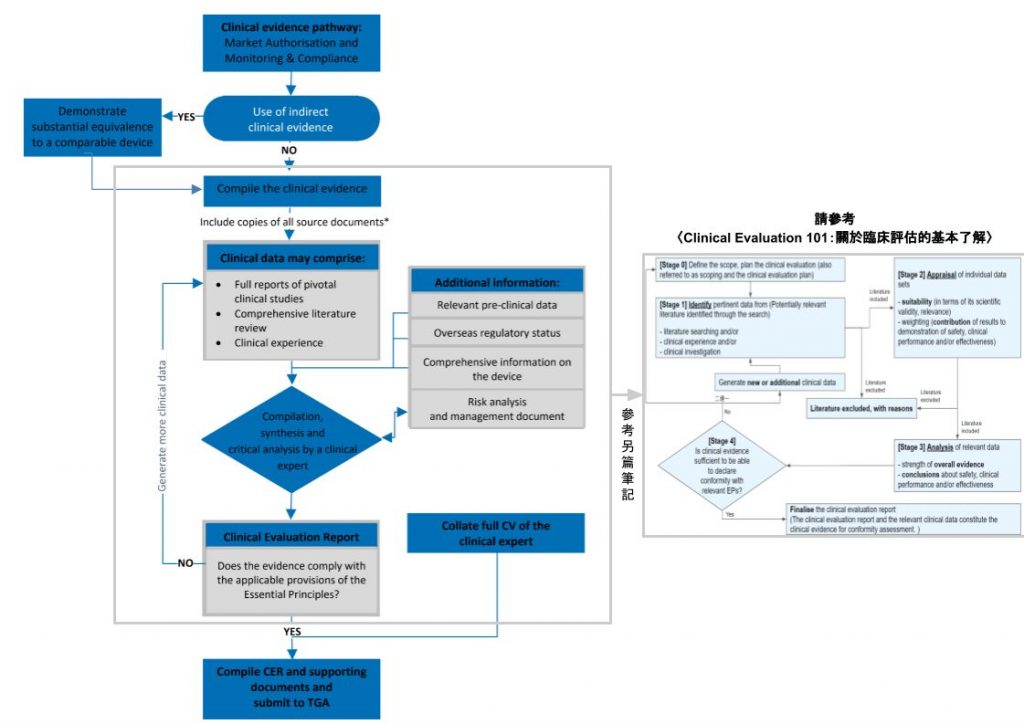
右圖則在〈CLINICAL EVALUATION 101:關於臨床評估的基本了解〉出現過,更細節地將 CER 的形成流程以四個階段呈現出來。
建議筆友可搭配兩圖來閱讀本筆記。
2. Clinical evidence 的用途與類別
Clinical data 經過 Clinical evaluation 後可生成 Clinical evidence,其目的是用來證明產品的 Benefit-risk profile 是可接受的,引用 TGA 的說法:
From this information, an acceptable benefit-risk profile may be demonstrated for a medical device, by showing that it performs as intended and that all identified undesirable effects and hazards, having been minimised during the design and development process, are outweighed by the benefits.
Clinical evidence guidelines for medical devices | Clinical evidence requirements (P.15)
| 什麼是 Clinical data 和 Clinical evidance?兩者的關係是什麼?更多相關細節,請參考〈Clinical Evaluation 101:關於臨床評估的基本了解〉的「4. Clinical data & Clinical evidence 是什麼?」 |
| 關於 Benefit-risk profile,請參考〈醫療器材上市前審查考量之利益與風險權衡要素 (Benefit-Risk evaluation)〉。 |
在 TGA 這份指引中,將 Clinical evidence 分為以下兩類:
- Direct clinical evidence – 由待評估醫材 (Subject device) 的 Clinical data 所產生的 Clinical evidence。
- Indirect clinical evidence – 由已證明實質相等 (Substantial Equivalence, SE) 的類似品的 Clinical data 所產生的 Clinical evidence。
| 這份 TGA 指引在 Part 2 的「Comparable devices including substantially equivalent devices」(P.47) 有討論「Comparable devices」、「Substantially equivalent devices」、「Predicates」三者差異,與 FDA、EU 的定義不同。 簡單來說,TGA 定義三者與產品本身 (Subject device) 的相似度由低至高排序為:Comparable devices < Substantially equivalent devices < Predicates。 而 TGA 指引說: 1) Substantially equivalent devices,「It may constitute the sole or major clinical data source for demonstrating compliance with the EPs.」,但 2) Comparable devices,「May form part of clinical evaluation and may support or supplement direct clinical data. It will not typically, in itself, constitute sufficient clinical evidence for the purpose of demonstrating compliance with the EPs – except for certain categories of lower risk devices….」 |
3. 識別 (Identification)
3.1 Search protocol
在前一筆記〈Clinical Evaluation 101:關於臨床評估的基本了解〉的 7.1 章節「Literature searching」裡提到:
在執行 Literature searching 前,須先建立 Literature search protocol,內容包含:
– [Source] 除說明資料來源外,還須解釋為何選擇這些來源;
– [Extent] 資料庫的搜索策略,會影響在特定資料來源內搜尋到多少文獻;
– [Selection criteria] 資料的納入/排除準則,並且須說明準則的設立原因;
– [Duplicated data addressing strategies] 處理不同文獻中重覆出現相同資料的策略。
TGA〈Clinical evidence guidelines for medical devices〉指引還規定 Search protocol 內要列出 (P.24):
- Search terms used (including key words and MeSH headings),
- Date searched,
- Period covered by the search,
- Search limits applied (including language, study design, etc.) and
- Inclusion and exclusion criteria.
3.1.1 PICO 手法 > 關鍵字
此份 TGA 指引進一步說明 (P.24),這階段可利用「Population, Intervention, Comparator(s) and Outcome(s) (PICO)」手法來聚焦問題。
以下解釋 PICO 四要素:
| 元素 | 英文全名 | 中文全名 | 解釋 (Ref: 成大護理系「實證護理步驟」) |
|---|---|---|---|
| P | Patients and/or Problem and/or Participants | 病人/母群體/問題/參與者 | – 某種疾病或狀況下的族群 (E.g., 血壓病人、泌尿道感染的病人、接受保護隔離的病人) – 某種基本資料下的族群 (E.g., 男性病人、具大學學歷的照顧者) – 某種環境 (E.g., 加護病房) |
| I | Intervention or Exposure | 介入 (處置) | 所關注的行動 (治療 / 介入 / 護理方式 / 衛教模式)、診斷工具、暴露因子、某種危險行為。 |
| C | Comparison / comparator(s) | 對照 | 既有或對照的行動、診斷工具、暴露因子、某種危險行為。 |
| O | Outcomes | 結果 | – 有意義可測量的臨床結果。例如,在「有效性」方面,可看改善症狀、增加存活率、減少感染率、避免危險因子;而關於「安全」,則死亡、副作用可作為 Outcomes。 – 可以是「生理指標」,像是體溫、血糖、疼痛指數、肺功能、 關節活動度…,或者是「社會心理指標」,例如情緒、行為、生活品質…。 |
例如:
| P | I | C | O | 範例來原 |
|---|---|---|---|---|
| 65 歲男性有中風及頸動脈狹窄病史 | 服用 Aspirin | 服用 Placebo (安慰劑) | 中風再發的機率 | 實證醫學入門 |
| (從「診斷」角度來發問) 疑似有五十肩患者 | 病人自我檢測表 | 醫師問診與理學檢查 | 診斷正確性 | 系統性文獻回顧研究之文獻搜尋方法 |
| (從「治療」角度來發問) 五十肩患者 | 肩關節囊擴張術 | 不做手術 | 肩關節活動程度 | Ibid |
| (從「傷害」角度來發問) 五十肩的中年男性 | 小針刀 | 一般針灸 | 神經損傷 | Ibid |
| (從「病因」角度來發問) 中年男性 | 奶爸史併高血糖 | 沒有高血壓 | 五十肩的發生 | Ibid |
| (從「篩檢」角度來發問) 有肝癌家族史的 中年男性 | 每年一次 完整肝功能篩檢 | 不做肝功能篩檢 | 肝癌發生率 | Ibid |
| 婦科手術病人 | 膀胱訓練 | 不做膀胱訓練 | 尿失禁機率 | 實證護理步驟 |
| 接受婦科手術後之病人 | 使用清水施行導尿管護理 | 使用優碘施行導尿管護理 | 泌尿道感染率 | Ibid |
| 加護病房 | 噪音管制教育計畫 | 沒有教育 | 降低環境音量效果 | Ibid |
| 50歲以上高血壓之中老年人 | 減重飲食 | 沒有減重飲食 | 血壓及中風發生率 | Ibid |
| 腎臟移植病人 | 生活經驗為何 | 居家環境下 | (這是質性問題,因此「I」是「Phenomena of Interest」,合併「CO」為「Context」。) | Ibid |
實際情況如下範例:

3.1.2 關鍵字 > 自由詞彙(同義字)
訂出四個 PICO 元素的關鍵字後,還要想想有哪些關鍵字衍伸出的自由詞彙 (同義字),例如同樣指禽流感,有人說「bird flu」,有人使用「avian flu」。
以下節錄自〈系統性文獻回顧研究之文獻搜尋方法〉的範例,以「Medication compliance」作為關鍵字為例,其同義字可由以下六個方向去發想:
| 六個延伸方向 | 同義字 |
|---|---|
| 縮寫 | MC |
| 情境上 | Noncompliance、Persistence |
| 廣狹義 | Furniture、Bed、Chair、Desk |
| 同義異形 | Adherence |
| 單複數/詞性/時態 | Compliable、Compliant、Compliancy、Complia* (切截字) |
| 英美式 | Edema、Oedema |

| 更多關於 PICO 的細節,可以參考陽明大學穆佩芬教授的「PICO」,裡面有很多範例以及說明。 |
3.2 Post-Market Data
在〈Clinical Evaluation 101:關於臨床評估的基本了解〉第 7 章節「Stage 1 – 資料識別 (Identification)」提到有三個管道可取得臨床評估的資料:
- Literature searching (文獻搜尋)、
- Clinical experience (臨床使用經驗,aka Real World Data),和
- Clinical investigation (臨床試驗)。
上面的 3.1 章節「Search protocol」所講的來源主要是「Literature searching」,而這章節則是在說明 TGA 指引對於第二個取得臨床評估資料的管道 –「(廣義,不一定有經同儕審查) Clinical experience」的分析建議 (P.25):
- 依「年」與「國家」或「區域」分析銷售量;
- 依「年」分析客訴發生的數量與類型,不只要統計確認是產品相關的抱怨數量,確認前的抱怨總數也要列入報告中;
- 依「年」分析不良事件 (含嚴重不良事件) 及警示事件 (Vigilance data),同樣要統計確認前/後與產品相關的事件數量,並且依「類型」(E.g. Device malfunction、Use error、Inadequate design 或 Manufacture) 與「結果 (Clinical outcome)」(E.g. Death、Amputation、Surgical procedure required、No harm to patient) 分門別類;
- 分析所有自願或強制的召回;以及
- 分析 Post-Market Clinical Follow-up (PMCF) 的資料。
告中要說明,上面這些資料是來自待評估醫材 (Subject device) 還是類似品。
Clinical experience 的其中一類資料 – Post-Market data,對於發現不常見但嚴重的事件,或者需經長時間使用才可得到的資訊是很有幫助的。
| TGA 指引詳細解釋了「Clinical experience data」、「RWD」、「RWE」的關係 (P.27): 1. Clinical experience data 是指「Data generated through any clinical use of the device that is not related to clinical investigation.」,包含 Post-market surveillance reports、Sales and complaints data、Vigilance reports、Clinically relevant field corrective safety actions; 2. Clinical experience data 的其它來源,例如 Electronic Health Records (EHRs)、Claims and billing activities、Product and disease registries、Patient-generated data including in home-use settings、Data gathered from other sources that can inform on health status (such as mobile devices) 可稱為 Real World Data (RWD); 3. RWD 經過分析即成 Real-world evidence (RWE),屬於 Clinical evidence 的一種。 另外,CDE 在第 95 期當代醫藥法規月刊〈伴同式體外診斷醫療器材之法規管理現況及趨勢〉一文中也解釋: 「RWE 係指非由臨床試驗所收集到的臨床使用證據。不同於真實世界數據 (real world data, RWD)。RWE 係使用有相關性 (relevance) 及可靠性 (reliability) 的 RWD 進行分析、確認後的結果。」 更多資訊可參考 FDA 指引〈Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices〉 |
| 請參考〈Clinical Evaluation 101〉第 7.2 章節「Clinical experience」,以了解 Clinical experience 的定義、和 Clinical investigation 的差異,以及來源。 |
4. 評讀 (Appraisal) – Selection strategy
| 以下是以 TGA 指引 的內容為主,但大家可另外參考: 1. Clinical Evaluation (MEDDEV 2.7/1,Rev. 4) 的「A5. Literature search and literature review protocol, key elements」(P.37)、「A6. Appraisal of clinical data」(P.40)、「A7. Analysis of the clinical data」(P.41); 2. Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019) 的「Appendix E: Some Examples to Assist with the Formulation of Criteria」、「Appendix F: A Possible Method of Appraisal」。 |
照著第 3 章節制訂出的 Search protocol 找到一堆文獻後,接著要評估這堆文獻的 Suitability (適用性) 與 Contribution (貢獻度),剔除一些沒用的資料。
這部分除了參考〈Clinical Evaluation 101:關於臨床評估的基本了解〉的第 8 章節「Stage 2 – 資料評讀 (Appraisal)」外,TGA 指引還建議要有流程圖 (Flow diagram) (P.24),以詳細展示篩選流程的每一步驟。例如,標示出每步驟納入/排除的數量、加權比重 (Weighting criteria) 的細節。
總之,要詳細到「Enable the clinical assessor to understand how the list of studies included in the review was compiled.」(TGA 指引,P.24)
4.1 初篩除找不到全文 & 不相關文獻
找到一堆資料後,如何初步篩選資料呢?可參考 PRISMA 去刪除重複、找不到完整報告、或不相關的文獻。

Part (A) 是用來展示「Database Search」的流程;Part (B) 則是為了展示「Grey Literature Search」的結果。後者是指「從 Database 以外管道而得到的文獻」,例如,從某文章的 Reference 或 Google Scholar 得到的。
圖/系統性文獻回顧研究之文獻搜尋方法
以下截錄 UNC Health Sciences Library「Creating a PRISMA flow diagram」關於 PRISMA flow diagram 的解說:
- 下載 PRISMA flow diagram 模板 (PRISMA Flow Diagram 官網)
- 依照第 3 章節「Search protocol」訂出的計畫開始在資料庫中檢索相關文獻,記錄各資料庫搜尋數量,以及加總所有文獻數量。
- 步驟 (1),排除重複 (Duplicates) 的文獻,並記錄排除數量。
- 步驟 (2),記錄無重複文獻數量 (總數 – 重複文獻數)。
- 步驟 (3),瀏覽文獻標題與摘要 (Titles and abstracts),排除不相關的文獻,並記錄排除數量。在此步驟,不強制一定要記錄排除原因。
- 步驟 (4),記錄剩餘的文獻總數 (總數 – 重複的數 – 標題/摘要與研究主題不相關的數)。
- 步驟 (5),排除那些無法找到全文 (Full text) 的文獻。
- 步驟 (6),記錄可以找到全文的文獻數量 (總數 – 重複的數量 – 標題/摘要與研究主題不相關的數量 – 無法找到全文的數量)。
- 步驟 (7),瀏覽全文,並刪除那些不恰當的文獻,例如,Wrong setting、Wrong patient population、Wrong intervention、Wrong dosage。在此階段,就要記錄排除原因了!(TGA 指引也如是說 (P.24))
- 步驟 (8),記錄經過層層篩選,最後留下的文獻總數 (Database Search + Grey Literature Search)。
| PRISMA 步驟 (7) 要我們刪除「不恰當的文獻」,但怎樣才算「不恰當」呢?大家可參考從以下方法中挑選適合的工具: 1) PRISMA 2020 Checklist (說明:PRISMA 2020 Explanation and Elaboration) 2) MOOSE (說明:Meta-analysis of Observational Studies in Epidemiology) 3) 第 4.2 章節「再評研究品質」內提及的各種評讀方法 |
4.2 再評研究品質
PRISMA flow diagram 篩選流程走到「步驟 (7)」時,有什麼方法篩選品質不佳的文件呢?
TGA 指引針對不同類型的實驗設計,推薦以下四個 Quality appraisal tools (aka critical appraisal tool, CAT),用來進一步評讀研究品質:
| Tool | Applicable study designs |
|---|---|
| Jadad Score | Randomised studies (Randomized clinical trials, RCT) |
| Downs & Black | Randomised & non-randomised studies |
| QUADAS | Studies of diagnostic accuracy |
| AMSTAR | Systematic reviews |
TGA 指引還另外推薦:Scottish Intercollegiate Guidelines Network (SIGN)、Centre for Evidence-Based Medicine (CEBM)、Cochrane Collaboration’s Handbook for Systematic Reviews of Interventions。(我…沒力氣去看了 Orz)
而 MEDDEV 2.7.1 (Rev.3) (現行版本為 Rev. 4) 也提供評估 Suitability 及 Data contribution 的簡易評讀方法:(簡單到缺少很多細節)
| Suitability Criteria | Description | Grading System |
|---|---|---|
| Appropriate device | Were the data generated from the device in question? | D1 Actual device D2 Comparable device D3 Other medical device |
| Appropriate device application | Was the device used for the same intended use (e.g. methods of deployment, application, etc.)? | A1 Same use A2 Minor deviation A3 Major deviation |
| Appropriate patient group | Were the data generated from a patient group that is representative of the intended treatment population (e.g. age, sex, etc.) and clinical conditions (i.e. disease, including state and severity)? | P1 Applicable P2 Limited P3 Different population |
| Acceptable report/data collation | Do the reports or collations of data contain sufficient information to be able to undertake a rational and objective assessment? | P1 High quality P2 Minor deficiencies P3 Insufficient information |
| Data Contribution Criteria | Description | Grading System |
|---|---|---|
| Data source type | Was the design of the study appropriate? | T1 Yes T2 No |
| Outcome measures | Do the outcome measures reported reflect the intended performance of the medical device? | O1 Yes O2 No |
| Follow up | Is the duration of follow-up long enough to assess treatment effects and identify complications? | F1 Yes F2 No |
| Statistical significance | as a statistical analysis of the data been provided and is it appropriate? | S1 Yes S2 No |
| Clinical significance | Was the magnitude of the treatment effect observed clinically significant? | C1 Yes C2 No |
4.2.1 Randomised studies (RCT) 評讀法 – Jadad Score
用於評讀 Randomized clinical trials (RCT) 文獻的 Jadad Score 方法,用來從試驗的 Randomization、Masking (Blinding)、Accountability (Withdrawals and dropouts) 此三大面向去鑑別一個 RCT 的試驗品質。
| Randomization | A method to generate the sequence of randomization will be regarded as appropriate if it allowed each study participant to have the same chance of receiving each intervention and the investigators could not predict which treatment was next. Methods of allocation using date of birth, date of admission, hospital numbers, or alternation should not be regarded as appropriate. |
| Double blinding | A study must be regarded as double blind if the word “double blind” is used. The method will be regarded as appropriate if it is stated that neither the person doing the assessments nor the study participant could identify the intervention being assessed, or if in the absence of such a statement the use of active placebos, identical placebos, or dummies is mentioned. |
| Withdrawals and dropouts | Participants who were included in the study but did not complete the observation period or who were not included in the analysis must be described. The number and the reasons for withdrawal in each group must be stated. If there were no withdrawals, it should be stated in the article. If there is no statement on withdrawals, this item must be given no points. |
上面三大重點解釋摘錄自〈A General Framework for the Evaluation of Clinical Trial Quality〉,下面的 Jadad Score 量表一樣摘錄自此篇文章:
| Item | Examples (Ref: Jadad scale for reporting randomized controlled trials) | Score |
|---|---|---|
| Was the study described as randomized (this includes words such as randomly, random, and randomization)? | “The patients were randomly assigned into two groups” | 0/+1 |
| Was the method used to generate the sequence of randomization described and appropriate (table of random numbers, computer-generated, etc)? | The randomization was accomplished using a computer-generated random number list, coin toss or well-shuffled envelopes | 0/+1 |
| Was the study described as double blind? | “The trial was conducted in a double-blind fashion” | 0/+1 |
| Was the method of double blinding described and appropriate (identical placebo, active placebo, dummy, etc)? | – Use of identical tablets or injectables, identical vials – Use of tablets with similar looks but different taste | 0/+1 |
| Was there a description of withdrawals and dropouts? (The fate of all patients in the trial is known. If there are no data the reason is stated.) | “There were 40 patients randomized but the data from 1 patient in the treatment group and 2 in the control were eliminated because of a break in protocol” | 0/+1 |
| Deduct one point if the method used to generate the sequence of randomization was described and it was inappropriate (patients were allocated alternately, or according to date of birth, hospital number, etc). | The group assignment was accomplished by alternate assignment, by birthday, hospital number or day of the week | 0/-1 |
| Deduct one point if the study was described as double blind but the method of blinding was inappropriate (e.g., comparison of tablet vs. injection with no double dummy) | Incomplete masking | 0/-1 |
用 Jadad Score 量表給予每篇研究一個品質分數後,可統整成如下結果:

作者們使用 Modified Jadad Scale 評定各篇研究設計的品質。Modified Jadad Scale 共 8 題,每題 1 分,總分為 0 ~ 8 分,分別為評定有無描述隨機化 (有 +1 分,無則 0 分)、隨機化是否適當 (適當 +1 分,未描述 0 分,不適當則 -1 分)、描述盲化 (雙盲 +1 分、單盲 +0.5 分,未描述 +0 分)、盲化是否適當 (適當 +1 分,未描述 0 分,不適當則 -1 分)、描述退出人數及理由、描述納入或排除標準、描述評估不良反應、描述統計分析方法 (有描述就 +1 分,無描述為 0 分)。分數愈高表示研究品質愈好。
| 多少分以下的研究才要被排除呢?Wikipedia 說:「A researcher conducting a systematic review for example might elect to exclude all papers on the topic with a Jadad score of 3 or less.」(Refer: Jadad scale) |
4.2.2 RCT & non-RCT studies 評讀法 – Downs & Black Checklist
Downs & Black checklist 分成以下五大類:
- Reporting – 十小項,用來評估文獻內所提供的資訊是否足以讓讀者對研究結果做出無偏誤 (Unbiased assessment) 的評估。
- External validity – 三小項,用來評估研究結果能有多大程度可以概括 (Generalised) 受試者 (Study subjects) 的族群。
- Internal validity (bias) – 七小項,目的是去評估介入措施 (Intervention) 與實驗結果 (Outcome) 的偏誤問題。
- Internal validity (confounding, selection bias) – 六小項,用此評估受試者選擇上的偏誤。
- Power – 只有一項,用來評估研究的負面結果是否為偶然產生的 (Due to chance)。
(上/下面的 Downs & Black 量表與解釋均摘錄自〈The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions〉;Paper 說「The total maximum score was therefore 31」但為啥我自己加總出的最高分是 32?)
4.2.2.1 Reporting
| Item | Brief Question (Ref: Downs and Black Checklist for Quality Assessment) | Criteria | Explanation | Score |
|---|---|---|---|---|
| 1 | Hypothesis/aim/objective clearly described | Is the hypothesis/aim/objective of the study clearly described? | – | Yes = 1; No = 0 |
| 2 | Main outcomes in Introduction or Methods | Are the main outcomes to be measured clearly described in the Introduction or Methods section? | If the main outcomes are first mentioned in the Results section, the question should be answered no. | Yes = 1; No = 0 |
| 3 | Patient characteristics clearly described | Are the characteristics of the patients included in the study clearly described ? | In cohort studies and trials, inclusion and/or exclusion criteria should be given. In case-control studies, a case-definition and the source for controls should be given. | Yes = 1; No = 0 |
| 4 | Interventions of interest clearly described | Are the interventions of interest clearly described? | Treatments and placebo (where relevant) that are to be compared should be clearly described. | Yes = 1; No = 0 |
| 5 | Principal confounders clearly described | Are the distributions of principal confounders in each group of subjects to be compared clearly described? | A list of principal confounders is provided. (這題在問什麼呢?請參考「Downs and Black checklist– question 5?」) | Yes = 2; Partially=1; No = 0 |
| 6 | Main findings clearly described | Are the main findings of the study clearly described? | Simple outcome data (including denominators and numerators) should be reported for all major findings so that the reader can check the major analyses and conclusions. (This question does not cover statistical tests which are considered below). | Yes = 1; No = 0 |
| 7 | Estimates of random variability provided for main outcomes | Does the study provide estimates of the random variability in the data for the main outcomes? | In non normally distributed data the inter-quartile range of results should be reported. In normally distributed data the standard error, standard deviation or confidence intervals should be reported. If the distribution of the data is not described, it must be assumed that the estimates used were appropriate and the question should be answered yes. | Yes = 1; No = 0 |
| 8 | All adverse events of intervention reported | Have all important adverse events that may be a consequence of the intervention been reported? | This should be answered yes if the study demonstrates that there was a comprehensive attempt to measure adverse events. (A list of possible adverse events is provided). | Yes = 1; No = 0 |
| 9 | Characteristics of patients lost to follow-up described | Have the characteristics of patients lost to follow-up been described? | This should be answered yes where there were no losses to follow-up or where losses to follow-up were so small that findings would be unaffected by their inclusion. This should be answered no where a study does not report the number of patients lost to follow-up. | Yes = 1; No = 0 |
| 10 | Probability values reported for main outcomes | Have actual probability values been reported(e.g. 0.035 rather than <0.05) for the main outcomes except where the probability value is less than 0.001? | – | Yes = 1; No = 0 |
4.2.2.2 External validity
| Item | Brief Question (Ref: Downs and Black Checklist for Quality Assessment) | Criteria | Explanation | Score |
|---|---|---|---|---|
| 11 | Subjects asked to participate were representative of source population | Were the subjects asked to participate in the study representative of the entire population from which they were recruited? | The study must identify the source population for patients and describe how the patients were selected. Patients would be representative if they comprised the entire source population, an unselected sample of consecutive patients, or a random sample. Random sampling is only feasible where a list of all members of the relevant population exists. Where a study does not report the proportion of the source population from which the patients are derived, the question should be answered as unable to determine. | Yes = 1; No = 0; unable to determine (UTD) = 0 |
| 12 | Subjects prepared to participate were representative of source population | Were those subjects who were prepared to participate representative of the entire population from which they were recruited? | The proportion of those asked who agreed should be stated. Validation that the sample was representative would include demonstrating that the distribution of the main confounding factors was the same in the study sample and the source population. | Yes = 1; No = 0; UTD = 0 |
| 13 | Location and delivery of study treatment was representative of source population | Were the staff, places, and facilities where the patients were treated, representative of the treatment the majority of patients receive? | For the question to be answered yes the study should demonstrate that the intervention was representative of that in use in the source population. The question should be answered no if, for example, the intervention was undertaken in a specialist centre unrepresentative of the hospitals most of the source population would attend. | Yes = 1; No = 0; UTD = 0 |
4.2.2.3 Internal validity – bias
| Item | Brief Question (Ref: Downs and Black Checklist for Quality Assessment) | Criteria | Explanation | Score |
|---|---|---|---|---|
| 14 | Study participants blinded to treatment | Was an attempt made to blind study subjects to the intervention they have received ? | For studies where the patients would have no way of knowing which intervention they received, this should be answered yes. | Yes = 1; No = 0; UTD = 0 |
| 15 | Blinded outcome assessment | Was an attempt made to blind those measuring the main outcomes of the intervention? | – | Yes = 1; No = 0; UTD = 0 |
| 16 | Any data dredging clearly described | If any of the results of the study were based on “data dredging”, was this made clear? | Any analyses that had not been planned at the outset of the study should be clearly indicated. If no retrospective unplanned subgroup analyses were reported, then answer yes. | Yes = 1; No = 0; UTD = 0 |
| 17 | Analyses adjust for differing lengths of follow-up | In trials and cohort studies, do the analyses adjust for different lengths of follow-up of patients, or in case-control studies, is the time period between the intervention and outcome the same for cases and controls ? | Where follow-up was the same for all study patients the answer should yes. If different lengths of follow-up were adjusted for by, for example, survival analysis the answer should be yes. Studies where differences in follow-up are ignored should be answered no. | Yes = 1; No = 0; unable to determine (UTD) = 0 |
| 18 | Appropriate statistical tests performed | Were the statistical tests used to assess the main outcomes appropriate? | The statistical techniques used must be appropriate to the data. For example nonparametric methods should be used for small sample sizes. Where little statistical analysis has been undertaken but where there is no evidence of bias, the question should be answered yes. If the distribution of the data (normal or not) is not described it must be assumed that the estimates used were appropriate and the question should be answered yes. | Yes = 1; No = 0; UTD = 0 |
| 19 | Compliance with interventions was reliable | Was compliance with the intervention/s reliable? | Where there was non compliance with the allocated treatment or where there was contamination of one group, the question should be answered no. For studies where the effect of any misclassification was likely to bias any association to the null, the question should be answered yes. | Yes = 1; No = 0; UTD = 0 |
| 20 | utcome measures were reliable and valid | Were the main outcome measures used accurate (valid and reliable)? | For studies where the outcome measures are clearly described, the question should be answered yes. For studies which refer to other work or that demonstrates the outcome measures are accurate, the question should be answered as yes. | Yes = 1; No = 0; UTD = 0 |
4.2.2.4 Internal validity – confounding (selection bias)
| Item | Brief Question (Ref: Downs and Black Checklist for Quality Assessment) | Criteria | Explanation | Score |
|---|---|---|---|---|
| 21 | All participants recruited from the same source population | Were the patients in different intervention groups (trials and cohort studies) or were the cases and controls (case-control studies) recruited from the same population? | For example, patients for all comparison groups should be selected from the same hospital. The question should be answered unable to determine for cohort and case-control studies where there is no information concerning the source of patients included in the study. | Yes = 1; No = 0; UTD = 0 |
| 22 | All participants recruited over the same time period | Were study subjects in different intervention groups (trials and cohort studies) or were the cases and controls (case-control studies) recruited over the same period of time? | For a study which does not specify the time period over which patients were recruited, the question should be answered as unable to determine. | Yes = 1; No = 0; UTD = 0 |
| 23 | Participants randomized to treatment(s) | Were study subjects randomised to intervention groups? | Studies which state that subjects were randomised should be answered yes except where method of randomisation would not ensure random allocation. For example alternate allocation would score no because it is predictable. | Yes = 1; No = 0; UTD = 0 |
| 24 | Allocation of treatment concealed from investigators and participants | Was the randomised intervention assignment concealed from both patients and health care staff until recruitment was complete and irrevocable? | All non-randomised studies should be answered no. If assignment was concealed from patients but not from staff, it should be answered no. | Yes = 1; No = 0; UTD = 0 |
| 25 | Adequate adjustment for confounding | Was there adequate adjustment for confounding in the analyses from which the main findings were drawn? | This question should be answered no for trials if: the main conclusions of the study were based on analyses of treatment rather than intention to treat; the distribution of known confounders in the different treatment groups was not described; or the distribution of known confounders differed between the treatment groups but was not taken into account in the analyses. In non-randomised studies if the effect of the main confounders was not investigated or confounding was demonstrated but no adjustment was made in the final analyses the question should be answered as no. | Yes = 1; No = 0; UTD = 0 |
| 26 | Losses to follow-up taken into account | Were losses of patients to follow-up taken into account? | If the numbers of patients lost to follow-up are not reported, the question should be answered as unable to determine. If the proportion lost to follow-up was too small to affect the main findings, the question should be answered yes. | Yes = 1; No = 0; unable to determine (UTD) = 0 |
4.2.2.5 Power
| Item | Brief Question (Ref: Downs and Black Checklist for Quality Assessment) | Criteria | Explanation | Score |
|---|---|---|---|---|
| 27 | Sufficient power to detect treatment effect at significance level of 0.05 | Did the study have sufficient power to detect a clinically important effect where the probability value for a difference being due to chance is less than 5%? | Sample sizes have been calculated to detect a difference of x% and y%. (這題該如何回答呢?請參考「How do I score the Power question on Downs & Black checklist for measuring quality?」及「Does anyone have specific guidance for how to interpret question 27 (power) of the Downs and Black (1998) checklist?」) | 請見下表 Size of smallest intervention group → Score |
| Size of smallest intervention group → Score |
|---|
| <n1 → 0;n1–n2 → 1;n3–n4 → 2;n5–n6 → 3;n7–n8 → 4;n8+ → 5 |
| 幾分才算低分要排除呢? 大家可參考〈Database of Abstracts of Reviews of Effects (DARE)〉:「…composed of 27 questions, with maximum possible score of 28 for randomised studies, and 25 for non-randomised studies; a score of 26 to 28 was considered excellent, 20 to 25 good, 15 to 19 fair and 14 or below was considered poor.」(但 Item 27 最高分可到 5,所以總分是 32 欸!) |
4.2.2.6 Case-control/Cohort study 是什麼?
Downs & Black Checklist 中常提到的 Case-control study & Cohort study,這兩者分別是什麼呢?
什麼是 Case-control study?
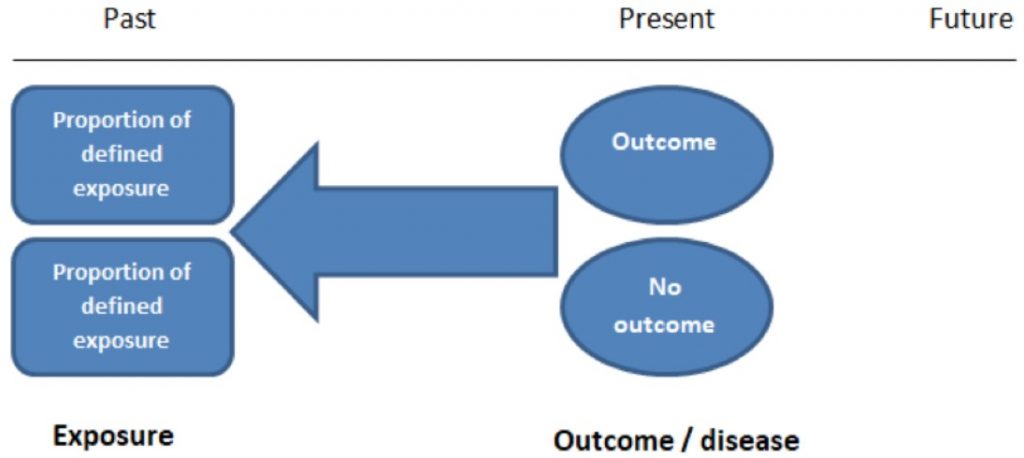
首先,要先明確定義出兩個族群,這兩個族群除了「Outcome/Disease」不一樣外 (一個族群有 Outcome,一個沒有),其餘特徵都要盡可能一樣。如下所說:
Case-control studies should include two groups that are identical EXCEPT for their outcome / disease status.
STUDENTS 4 BEST EVIDENCE | Case-control and Cohort studies: A brief overview
例如,研究心肌梗塞病因時,選擇 100 名心肌梗塞患者作為 A 族群,另選擇 100 名沒有發生過心肌梗塞的病患作為 B 族群,兩族群最好除了有/無發作過心肌梗塞外,其餘特徵越相近越好。
接著透過回溯 (Retrospective) 研究,看看是不是因為 A 族群在過去有暴露於某些風險因子導致其心肌梗塞。
Case-control study 最終的結果會是 Odds ratio (OR,勝算比)。
例如上述心肌梗塞的例子,若 A 族群中有 40 人患高血壓,而 B 族群中有 20 人患高血壓,則 OR = (40/60)/(20/80) = 2.67,代表患高血壓的人發生心肌梗塞的風險是沒有高血壓的人的 2.67 倍。(Ref: OR值的含义与解释)
以下是 Case-control study 的優缺點:
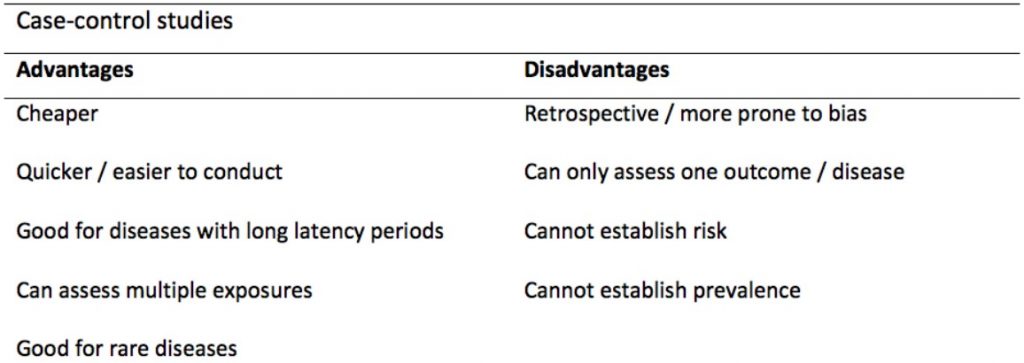
因為需要受試者回想自己曾暴露於哪些因子中,造成此類研究法其中一個主要的 Bias 就是「Recall bias」。例如,相較於健康正常產下小孩的母親,死胎 (Stillbirth) 的母親對於自己曾暴露於哪些因子的記憶可能更為深刻。
圖/Case-control and Cohort studies: A brief overview
什麼是 Cohort study?

和 Case-control study 不同的是,Cohort study 招募的受試者是:
Cohort studies should include two groups that are identical EXCEPT for their exposure status.
STUDENTS 4 BEST EVIDENCE | Case-control and Cohort studies: A brief overview
因此,無論有/無暴露於特定因子的兩族群受試者 (Exposed/Unexposed groups),都應該是從同一群體中招募來的 (Same source population)。接著就追蹤 (Follow up) 受試者一段時間,看看發展成特定 Outcome/Disease 的比例。
Cohort study 要先招募受試者,接著追蹤其一段時間,這有一個最大的問題就是「Attrition (流失/損耗)」。受試者可能因為種種原因,例如死亡,而無法繼續被追蹤,而導致「損耗性偏誤 (Attrition bias)」。
Cohort study 最終的結果會是 Risk ratio / Relative risk (RR)。
例如,想要知道心血管疾病 (CVD) 與高血壓 (HYPERTENSION) 的關係,可將其結果放入以下的 2 X 2 表中:

依上表計算,同時有高血壓與 CVD 的發生率為:a/(a+b) = 40/(40+81) = 33.06%;而只有 CVD 沒有高血壓的發生率為: c/(c+d) = 11/(11+834) = 1.30%。進一步可算出 RR = 33.06%/1.30% = 25.39。(Ref: R 軟體資料分析應用:相對風險、勝算比與邏輯斯迴歸分析)
依照慈濟醫學中心的〈醫學研究中常見的統計應用及誤用〉,RR 及 OQ (Odds ratio) 意義如下:
| 數值 | 代表意義 |
|---|---|
| RR 或 OR = 1 | 無論有無暴露於危險因子中,發生不良結果的可能性一樣。 |
| RR 或 OR > 1 | 暴露於危險因子中導致不良結果的風險增加。 (Common Rule:病例對照研究(Case-control study) 偏誤 (Bias) 較多,當 OR > 4 時較有意義;世代研究 (Cohort study) 較嚴謹,但仍有偏誤存在,故 RR > 3 時較有意義。) |
| RR 或 OR < 1 | 暴露於危險因子者比未暴露更不可能發生不良結果 |
以下是 Case-control study 的優缺點:

4.2.3 Studies of diagnostic accuracy 評讀法 – QUADAS-2
QUADAS 的全名是 Quality Assessment of Diagnostic Accuracy Studies,由 Penny Whiting 在 2003 年推出第一代,現在已修訂為第二代 – QUADAS-2。
QUADAS-2 透過以下四大關鍵領域 (Domain):(Ref: QUADAS-2)
- PATIENT SELECTION (病例選擇)、
- INDEX TEST (待評讀試驗)、
- REFERENCE STANDARD (黃金標準),及
- FLOW AND TIMING (疾病流程與進展),
來評估原始研究的 Risk of bias (RoB,偏誤風險) 和 Applicability (適用性,適用於待評讀問題的程度)。
若在上述四大關鍵領域的 Signaling questions 中,所有答案均為「Yes」,則 RoB 可評定為「Low」;若有任一個 Signaling question 答案是「No」,RoB 則為「High」。(Ref: 想写诊断meta分析,那你得了解QUADAS-2!;偏倚风险评估系列:(六)诊断试验)
4.2.3.1 執行流程
Stage 1 – 確認問題
可參考第 3.1.1 章節的「PICO 手法」來確認待評讀問題的明確內容。
Stage 2 – 根據需求建立/調整 QUADAS-2 評鑑表
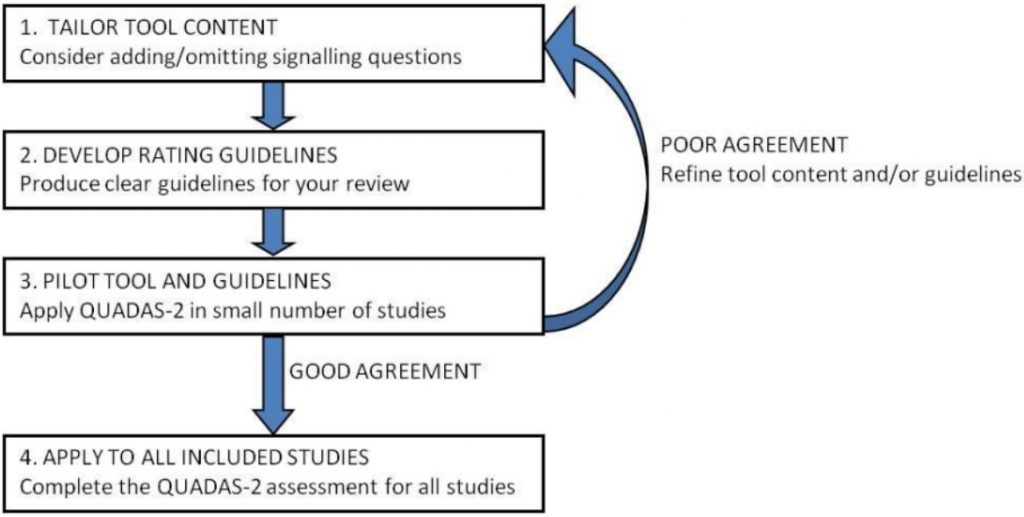
原始 QUADAS-2 評鑑表可依據評估需求增減 Signaling questions,但要避免增加太多使得評鑑過於複雜。無論增減,都須註明理由。
接著,至少由兩位評估者使用調整好的 QUADAS-2 去預評少數原始研究,如果一致性好,則可進入下一階段;若一致性不好,就要再次修改 QUADAS-2 評鑑表。
Stage 3 – 為每個原始研究畫流程圖
若原始研究中已有詳細流程圖,可以直接使用;若無或不足,則評估者可自行為每個原始研究繪製流程圖,但這部分不強制作為 QUADAS-2 的一部分。
Stage 4 – 透過 QUADAS-2 開始評估 Risk of bias (RoB) 及 Applicability
使用 QUADAS-2 最終並不會有一個總結性的「品質分數」,但結果報告中可交代以下內容:
- 總的 RoB – 如果原始研究都判定為「Low」,那總體即可評為「低偏誤風險」;若有一個或多個判定為「High」或「Unclear」,則可結論為「存在偏誤風險」。
- 整理每個關鍵領域偏誤風險為低、高、不清楚的原始研究數量。
- 討論 Signaling questions 發現的問題。
- 討論 Applicability。
請看以下對於 QUADAS-2 評鑑內容的解釋。
4.2.3.2 PATIENT SELECTION (病例選擇)
這個項目是用來評估「病例選擇方式是否有 Bias?」
| Description (記錄用來支持判斷 RoB 的資訊,目的是使評鑑過程透明化,並方便評估者之間討論) | Signaling questions (Yes/No/Unclear) | Risk of bias (RoB) (High/Low/Unclear) | Concerns regarding applicability (High/low/unclear) (適用性評估要和待評讀問題結合起來一起評估,不屬於 RoB,須於正式開始評估前決定要不要進行,以避免選擇性報告結果) |
|---|---|---|---|
| – Describe methods of patient selection – Describe included patients (prior testing, presentation, intended use of index test and setting) | 1. Was a consecutive or random sample of patients enrolled? (是否納入連續或隨機的病例?) 理想的研究應該是連續或隨機納入患有疑似疾病、符合條件的病患,以免發生 Bias。 2. Was a case-control design avoided? (是否避免病例對照研究?) 因為是事先已選定兩組對照,一組為「確診的試驗組」,另一組是「非確診的對照組」,事先知道了分組,所以很可能會誇大診斷的準確性。(可參考前面章節的「什麼是 Case-control study?」) 3. Did the study avoid inappropriate exclusions? (是否避免不恰當的排除?) 如果排除了難以診斷 (Difficult-to-diagnose) 的患者,則可能會高估診斷的正確性。 一般來說,納入的病例中,若難以解釋/不確定的結果佔 20% 以上,就可以評為「Yes」;如果所有結果都可以明確診斷,也就是沒有包含診斷不明確的病例,就應評為「No」;如果納入的病例中,難以解釋的結果的比例為 > 0% 且 < 20%,就可認定是「Unclear」。 | Could the selection of patients have introduced bias? | Are there concerns that the included patients do not match the review question? (納入的病患是否有不匹配待評讀問題的疑慮?) 原始研究中納入的病例與待評讀問題中的目標族群是否在某些面有不匹配的情況 (E.g., 疾病的嚴重程度、併發症、先前的檢查方法…)。 例如,相比小面積心肌梗塞,大面積的心肌梗塞在急性期通常會有較多的心肌酶與心肌肌鈣蛋白,進而提高靈敏度的估計。 |
4.2.3.3 INDEX TEST (待評讀試驗)
這個項目是用來評估「待評讀試驗的實施或解釋是否會產生 Bias?」
| Description | Signaling questions (Yes/No/Unclear) | Risk of bias (RoB) (High/Low/Unclear) | Concerns regarding applicability (High/Low/Unclear) |
|---|---|---|---|
| Describe the index test and how it was conducted and interpreted | 1. Were the index test results interpreted without knowledge of the results of the reference standard? (待評讀試驗的結果判讀是否在不知曉黃金標準試驗結果的情況下進行的?) 若已經知道黃金標準的結果,可能會影響待評讀試驗的解釋。 2. If a threshold was used, was it pre-specified? (若設定了閾值,那它是否是預先確定好的?) 若在結果判定前就訂出閾值,則為「Yes」,反之則「No」。 因為,如果研究中選擇的閾值是根據敏感度和/或特異度而選擇的最佳結果,那就很可能會高估診斷的正確性。 | Could the conduct or interpretation of the index test have introduced bias? | Are there concerns that the index test, its conduct, or interpretation differ from the review question? (待評讀試驗的實施或解釋是否有不匹配待評讀問題的疑慮?) |
4.2.3.4 REFERENCE STANDARD (黃金標準)
這個項目是用來評估「黃金標準的實施或解釋是否會產生 Bias?」
| Description | Signaling questions (Yes/No/Unclear) | Risk of bias (RoB) (High/Low/Unclear) | Concerns regarding applicability (High/Low/Unclear) |
|---|---|---|---|
| Describe the reference standard and how it was conducted and interpreted | 1. Is the reference standard likely to correctly classify the target condition? (黃金標準是否可正確區別目標狀態?) 因為判斷診斷是否正確,就是假設黃金標準的靈敏度和特異度都為 100%,再看待評讀試驗結果與黃金標準結果的差異,因此黃金標準很重要。 2. Were the reference standard results interpreted without knowledge of the results of the index test? (黃金標準結果的判讀是在不知道待評讀試驗結果下進行的嗎?) | Could the reference standard, its conduct, or its interpretation have introduced bias? | Are there concerns that the target condition as defined by the reference standard does not match the review question? (是否有黃金標準的目標疾病與待評讀問題不匹配的顧慮?) 這邊要考慮兩點:1) 文獻中的黃金標準是否與待評讀問題中所定義的納入研究的黃金標準一樣;2) 兩者對於目標疾病的定義是否相同 (E.g., 是否採用相同閾值判斷患者與非患者)。 |
4.2.3.5 FLOW AND TIMING (流程與進展)
這個項目是用來評估「流程與進展是否會產生 Bias?」
| Description | Signaling questions (Yes/No/Unclear) | Risk of bias (RoB) (High/Low/Unclear) |
|---|---|---|
| – Describe any patients who did not receive the index test(s) and/or reference standard or who were excluded from the 2×2 table (refer to flow diagram) – Describe the time interval and any interventions between index test(s) and reference standard | 1. Was there an appropriate interval between index test(s) and reference standard? (待評讀試驗與黃金標準試驗之間是否有適當的時間間隔?) 時間間格越短越好,因為間隔時間越長,疾病的康復/惡化都可能會造成結果判斷錯誤。 時間間格長一點,對慢性病的影響可能不大,但若是急性疾病,短期內的變化可能會影響判斷。 最理想的狀況是,同時對同一患者進行待評讀試驗與黃金標準試驗。 適當的時間間隔應該在正式使用 QUADAS-2 前就確定。 2. Did all patients receive a reference standard? (所有患者是否只接受一個黃金標準試驗?) 3. Did all patients receive the same reference standard? (所有患者都接受相同的黃金標準試驗?) 若 Q2 & Q3 是「否」,則可能會發生「驗證誤差 (Verification bias)」。 如果待評讀試驗的結果影響了是否使用黃金標準診斷,或者使用哪個黃金標準診斷,那麼診斷準確性的評估可能會發生 Bias。 例如,在「高敏感度心肌肌鈣蛋白檢測排除急性心肌梗死」的例子中,如果對陽性者進一步進行標準的肌鈣蛋白檢測和心電圖檢查 (黃金標準 1),而對陰性者透過臨床隨訪確定是否發生心肌梗死 (黃金標準 2),如此一來,就可能會將診斷試驗的假陰性結果錯分為真陰性,因為臨床隨訪可能會漏掉那些診斷試驗結果為陰性的急性心肌梗死患者,從而高估「高敏感度心肌肌鈣蛋白檢測排除急性心肌梗死」的正確性。 4. Were all patients included in the analysis? (是否所有的病例都納入分析?) | Could the patient flow have introduced bias? (流程是否造成 Bias?) |
4.2.3.6 QUADAS-2 範例
請參考〈中華流行病學雜誌〉的「偏倚风险评估系列:(六)诊断试验」。
4.2.4 Systematic reviews 評讀法 – AMSTAR
2007 年制定了第一版 AMSTAR (A Measure Tool to Assess Systematic Reviews),適用於評讀 Systematic reviews (SR) 的質量,並在 2017 年 9 月推出了 AMSTAR 2。
以下列出 AMSTAR 2 的十六項評估內容,並從〈系统评价方法学质量评价工具AMSTAR 2解读〉、〈AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both〉及相關附件摘錄重點。
十六項評估內容中,AMSTAR 2 作者們說以下七項尤為重要,稱為「AMSTAR 2 critical domains」:
對應不同類型的研究,可能 Critical domains 會有所變化。例如,Risk of bias (RoB) 對於設計嚴謹的 RCT 比較不重要;而在評估 Meta-analysis 的品質時,Item 4、7、15 就不是那麼關鍵了。
下面是 AMSTAR 2 作者們建議的整體信賴度評級 (Rating overall confidence):
| Rating | Results |
|---|---|
| High | o or one non-critical weakness: the systematic review provides an accurate and comprehensive summary of the results of the available studies that address the question of interest. |
| Moderate | More than one non-critical weakness: the systematic review has more than one weakness but no critical flaws. It may provide an accurate summary of the results of the available studies that were included in the review. (Multiple non-critical weaknesses may diminish confidence in the review and it may be appropriate to move the overall appraisal down from moderate to low confidence.) |
| Low | One critical flaw with or without non-critical weaknesses: the review has a critical flaw and may not provide an accurate and comprehensive summary of the available studies that address the question of interest. |
| Critically low | More than one critical flaw with or without non-critical weaknesses: the review has more than one critical flaw and should not be relied on to provide an accurate and comprehensive summary of the available studies. |
Item 1. Did the research questions and inclusion criteria for the review include the components of PICO?
作者應訂定明確具體的 PICO,以確保 Systematic reviews (SR) 納入正確適當的研究結果。(不一定有用 PICO 表格,只要有在文內明訂出 PICO 要素都沒問題。)
除了 PICO 外,視情況可能還要定義「Timeframe」(例如,當某些處置 (Intervention) 的效果只會在幾年後被觀察到時)。
| 詳細內容請參考第 3.1.1 章節的「PICO 手法」。 |
Item 2. Did the report of the review contain an explicit statement that the review methods were established prior to the conduct of the review and did the report justify any significant deviations from the protocol?
作者須先制定好 Systematic reviews (SR) 計畫,接著再跟著計畫執行 SR。若只有符合上述要求,那只算是「Partial Yes」。
如果執行 SR 前,計畫有經「Independent verification」,且後續也有照著原定計畫執行 SR,即可判定為「Yes」。Independent verification 的方法有 Registration (E.g., PROSPERO)、公開發表期刊 (E.g., BMJ Open) 或某些科研辦公室、試驗倫理委員會等等。
若實際執行與原定計畫有偏誤時,作者須解釋原因。
Item 3. Did the review authors explain their selection of the study designs for inclusion in the review?
不一定 Randomised controlled trials (RCT) 就比較好,某些主題的研究可能只能用 Non-randomized studies of interventions (NRSI;若沒處置 (I),則為 NRS為 NRS),例如,政策調整的影響 (Effects of policy changes)。
重點是,無論納入什麼研究方法 (Study types),作者都要解釋納入這些實驗設計到 SR 的原因。
同樣地,若針對同一處置,同時有 RCT 和 NRSI (或 NRS) 兩種研究方法去評估其效果,但作者在 SR 中只採用 RCT 的原始實驗,那作者也需要解釋如此作法對於評估結果並不會有不良影響 (E.g., incomplete summary of the effects of a treatment)。
Item 4. Did the review authors use a comprehensive literature search strategy?
作者至少應檢索兩種資料庫 (Bibliographic databases)。
報告內至少須包含「年份」與「資料庫」、「關鍵字」及/或「MESH terms」,還有「檢索策略」。
檢索結果最好還有用其他的資料來源補充,例如,Published reviews、Specialized registers、特定領域專家的建議、納入檢索的文獻的參考文獻。
此外,應檢索相關語言的資料,若有語言限制,也應解釋。
有時,Gray literature 有其重要性。例如,在評估政策時,某些 (非) 政府組織的報告只能從網路取得。這類資訊可能可從各大學網站、ResearchGate,甚至直接從研究者或公司等來源獲得。
Item 5. Did the review authors perform study selection in duplicate?
以 PRISMA 篩選文獻流程來說,在步驟 (7) 的「瀏覽全文,並刪除那些不恰當的文獻」時,較有可能因為主觀認知導致不同的篩選結果。
因此,AMSTAR 2 要求文獻篩選須有再現性。所以,至少要由兩名人員分別篩選文獻。第二名人員只要「Checked agreement on a sample of representative studies」即可。
若篩選意見不一致時,應透過共識流程 (Consensus process) 來取得共識。(兩人可試評 10% 文章,) 一致性的 Kappa 相關係數應 ≥ 0.80。
如果 SR 的作者有說明上面這些要求 (就是 Inter-rater agreement),那這題就可回答「Yes」。
Item 6. Did the review authors perform data extraction in duplicate?
經過上一題「文獻篩選」確定納入原始研究後,Systematic reviews (SR) 的作者們就要開始「提取數據」。
因為一份研究中可能會有多個治療效果 (Treatment effects),為了要選擇最符合 PICO 的資料,這個階段可能會產生偏誤。所以,與 Item 5 一樣要有再現性,兩名人員各自的數據提取結果的 Kappa 相關係數應 ≥ 0.80。
| 關於「提取數據」解釋,可參考〈Cochrane Handbook〉的「Chapter 5: Collecting data」。 |
Item 7. Did the review authors provide a list of excluded studies and justify the exclusions?
SR 作者須提供排除文獻清單,並說明排除原因。
排除理由可能有:Inappropriate/ irrelevant populations、Interventions、Controls…。但不可因為 RoB 而排除,因 RoB 是在完成文獻篩選、納入文獻後進行分析時才進行的 (Exclusion should not be based on risk of bias, which is dealt with separately and later in the review process (Ref: AMSTAR 2 guidance document))。
Item 8. Did the review authors describe the included studies in adequate detail?
應對納入的研究在 Research designs、Study populations、Interventions、Comparators、Controls、Outcomes、Analysis 等方面有詳細的描述。
上述資訊若描述夠清楚的話,還可協助判斷是否異質性 (Heterogeneity, e.g. by dose, age range, clinical setting etc.) 會影響處置結果 (Intervention effects)。
Item 9. Did the review authors use a satisfactory technique for assessing the risk of bias (RoB) in individual studies that were included in the review?
應對每一個納入 SR 的原始研究採用適當的方法去評讀其 RoB。
何謂「適當方法」?
在〈AMSTAR 2〉附件「AMSTAR 2 guidance document」的 Item 9 舉例:Newcastle Ottawa Scale (NOS)、SIGN、Mixed Methods Appraisal Tool (MMAT) 等都屬於適當方法,其中最廣為應用的是 Cochrane ROBINS-I。(我想,本筆記 第 4.2 章節 列出的方法也算吧。)
有哪些常見的偏誤呢?在上述附件中提到以下最常見的四項偏誤:
- Confounding – 例如,吸菸與飲酒此二因子會互相干擾;
- Sample selection bias – 例如,不應招募去戒菸所的人來研究吸菸與心臟病的關聯;
- Bias in measurement of exposures and outcomes – 例如,以「Exposure」來說,死胎 (Stillbirth) 的母親對於自己曾暴露於哪些因子的記憶可能更為深刻;以「Outcome」來說,選擇因腿部腫脹疼痛而常去醫院做超音波檢查的婦女為對象,研究深部靜脈栓塞 (Deep Vein Thrombosis) 與口服避孕藥的關係,這樣可能會有偏誤;
- Selective reporting of outcomes and analyses – 大規模的觀察性研究可能會從 Population databases 中分析出許多 Outcomes,若沒有預先定義好 Outcomes,研究者可能分析多種結果,並從中挑選那些暴露/不暴露有顯著差異的 Outcomes。同樣地,若沒事先定義分析方法,研究者也可能會挑選那些會分析出重大統計差異的方法。
Item 10. Did the review authors report on the sources of funding for the studies included in the review?
要註明資金來源,以使評估者了解此研究是 Commercially funded 或 Independently funded studies。
Item 11. If meta-analysis was performed did the review authors use appropriate methods for statistical combination of results?
是否有採用適當的統合分析方式?
例如,計畫書內是否有定義清楚研究主題?納入/排除條件是否有先定義好?是用固定式模型 (Fixed-effects model),或是隨機式模型 (Random-effects model)?有沒有寫清楚如何分析個別實驗之間的異質性 (Heterogeneity)?
| 關於 Meta-analysis (統合分析) 的介紹,可參考 CDE〈統合分析 (Meta-analysis) 簡介〉與台灣醫界於 2011 年出刊的〈臨床醫師如何閱讀統合分析(Meta-analysis)的論文〉。 |
Item 12. If meta-analysis was performed, did the review authors assess the potential impact of RoB in individual studies on the results of the meta-analysis or other evidence synthesis?
要評估統合分析報告中,作者是否有考慮,納入分析的個別研究的 RoB (偏誤風險) 對分析結果或其他證據的影響。
若作者僅有納入高品質的 RCT,可能這方面的討論會比較少;若有納入不同品質的 RCT,或者納入 Non-randomized studies (NRS,或 Non-randomized studies of interventions (NRSI)),則作者就要好好評估 RoB 對分析結果的影響。
Item 13. Did the review authors account for RoB in individual studies when interpreting/ discussing the results of the review?
無論是否有做統合分析 (Meta-analysis),作者都要解釋/討論所納入研究的 RoB,說明是否這些 RoB 會影響臨床照護政策 (clinical care or policy)。
這個討論很重要,尤其當納入不同程度 RoB 的 RCT 以及 NRSI 時更需要說明。
Item 14. Did the review authors provide a satisfactory explanation for, and discussion of, any heterogeneity observed in the results of the review?
作者須驗證異質性 (Heterogeneity) 存在的可能性,分析其可能的成因,並討論異質性對研究結論與建議的影響。
例如,Different study designs、Different methods of analysis、Different populations、Differing intensities of the intervention(s) (以藥品來說,就是 Dosages),以及 Item 1 的 PICO 要素、Item 9 提到的 RoB 都很有可能導致異質性。
Item 15. If they performed quantitative synthesis did the review authors carry out an adequate investigation of publication bias (small study bias) and discuss its likely impact on the results of the review?
若有定量分析時,應詳細研究 Publication bias (PB,出版性偏誤),並討論其對於資料解讀與結果討論的可能影響。
| 所謂的出版偏誤,即爲在大多數狀況下,研究人員總會傾向發表正向效果的文章,而負向效果的文章則不加以發表;就另一方面而言,期刋的編輯也傾向接受有正向效果的文章,而造成具負向效果或無明顯效果的文章不易爲人所知。這就形成了在收集文獻時,某些應存在的研究結果不易被發現。(Ref: 以斯帖統計顧問公司「統合分析(Meta-analysis):出版偏誤(publication bias)問題」) Publication bias can arise from a researcher’s decision not to publish negative results due to a combination of personal, financial, and professional pressures to publish ‘exciting’ results. (Ref: The University of Sydney「Publication bias: Why null results are not necessarily ‘dull’ results」) |
例如,由紅酒公司贊助所進行的「紅酒中的抗氧化劑對心血管的影響」研究,其 PB 很大機率會大於由獨立單位進行的研究。一般來說,可透過漏斗圖 (Funnel plot) 看出有無 PB。
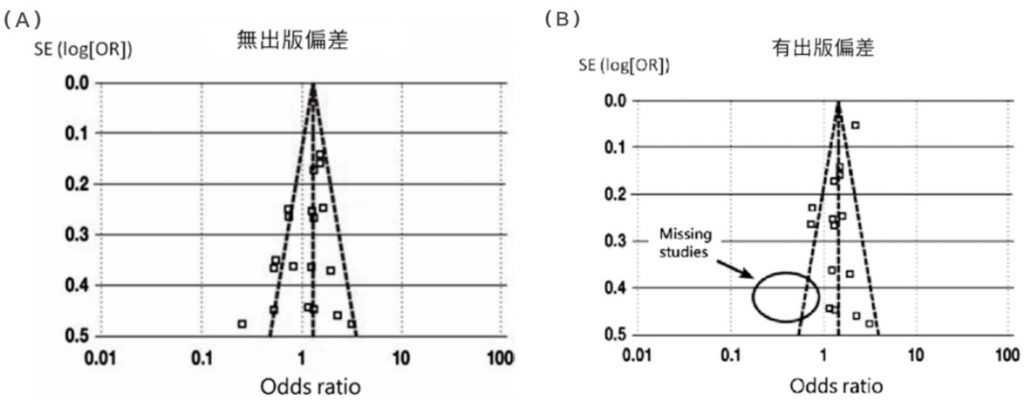
圖/臨床醫師如何閱讀統合分析 (Meta-analysis) 的論文 (台灣醫界, 2011, Vol. 54, No. 2)
或者也可透過 Sensitivity analyses,也就是執行兩次 Meta-analysis,一次納入所有研究去分析,另一次則刪除品質不佳的研究再分析,以確定「Are the findings robust to the decisions made in the process of obtaining them?」
若「When sensitivity analyses show that the overall result and conclusions are not affected by the different decisions that could be made during the review process」,則表示「The results of the review can be regarded with a higher degree of certainty.」
但如果「Where sensitivity analyses identify particular decisions or missing information that greatly influence the findings of the review」,就要「Greater resources can be deployed to try and resolve uncertainties and obtain extra information…. If this cannot be achieved, the results must be interpreted with an appropriate degree of caution.」(Ref:〈Cochrane Handbook for SR of Interventions〉的「10.14 Sensitivity analyses」)
Item 16. Did the review authors report any potential sources of conflict of interest, including any funding they received for conducting the review?
作者要報告任何潛在的利益衝突。
和 Item 10 不同的是,Item 16 特別提到「Authors have ties to companies that manufacture products included in the systematic review」,也就是說,除了資金贊助,還要考慮是否研究中有用到利益衝突的產品。
4.2.5 影響科學效力的範例
MEDDEV 2.7/1 (Rev. 4) 在附錄六「A6. Appraisal of clinical data」提供多個常見影響 Scientific validity 的範例。
4. Identification / Screening / Eligibility / Included flow

圖/Systematic review of the methodological quality and outcome measures utilized in exercise interventions for adults with spinal cord injury
5. 分析 (Analysis) & 結論
| 以下是以 TGA 指引 的內容為主,但大家可另外參考: 1. Clinical Evaluation (MEDDEV 2.7/1,Rev. 4) 的「A5. Literature search and literature review protocol, key elements」(P.37)、「A6. Appraisal of clinical data」(P.40)、「A7. Analysis of the clinical data」(P.41); 2. Clinical Evaluation (IMDRF MDCE WG/N56FINAL:2019) 的「Appendix E: Some Examples to Assist with the Formulation of Criteria」、「Appendix F: A Possible Method of Appraisal」。 |
關於「資料分析」,可參考前一筆記〈Clinical Evaluation 101:關於臨床評估的基本了解〉的第 9 章節「Stage 3 – 資料分析 (Analysis)」。
除了〈Clinical Evaluation 101〉的「9.2 Analysis 重點」外,TGA 指引建議 (P.24) 用「表格」整理各文獻的結果與研究特點,表格內容要包含所有結果。例如,樣本數 (Effect size estimate)、信賴區間 (Confidence interval)、不良事件發生率 (Adverse event rates for different types of adverse events) 等等。
另外關於分析 (Analysis) 與結論,除了前篇筆記的「9.3 Analysis 結論」外,TGA 指引也補充說:這部分不只是
This is not a simple summary of the individual study results, but a critique and discussion of the study method, results and outcomes and how these apply to the device when used for its intended purpose.
Review and critical analysis (P.24)
| 雖然常常會做出「只是再次統整一次 Data 後,寫『所以我們產品很安全有效啦』的聲明」的 CER,但 TGA 指引 (P.38) 說這樣是不足夠的: 「It is critical that a CER (which serves to detail the clinical evidence as required by the legislation) is not simply a summary of the data, followed by a statement that the data demonstrate safety and performance. This approach does not represent an adequate clinical evaluation.」 |
而此階段 (Analysis) 的目標是
To make a benefit-risk determination regarding whether the appraised data sets available for a medical device collectively demonstrate the safety, clinical performance and/or effectiveness of the device in relation to its intended use.
Analysis of the clinical data (P.33)
| 更多關於「Benefit-risk determination」的細節,請參考另篇筆記〈醫療器材上市前審查考量之利益與風險權衡要素 (Benefit-Risk evaluation)〉。 |
| 本階段重點在於確認「Clinical evidence」、「Intended purpose」、「Indications」、「Contraindications」、「Earnings」、「Precautions」與「Actual and potential adverse effects」是否一致。(TGA 指引,P.38) |
TGA 指引說,此階段應考慮 (但不應侷限於) 以下幾點:
- the strengths and limitations of the clinical data presented in support of the safety and performance of the device for the intended purpose(s) e.g. level and nature of evidence, bias, confounders, length of follow-up (臨床資料用來支持安全與功效的強度與限制)
| 什麼是 Clinical data 和 Clinical evidance?兩者的關係是什麼?更多相關細節,請參考〈Clinical Evaluation 101:關於臨床評估的基本了解〉的「4. Clinical data & Clinical evidence 是什麼?」 |
- the clinical significance of the benefits of the device for the intended purpose(s) as demonstrated by the clinical data (臨床資料呈現出對目標族群所造成的利益的臨床意義)
- based on the clinical data provided and on a sound statistical approach, a reasonable prediction of the proportion of ’responders’ out of the target group or subgroups should be made (透過合理統計臨床資料後,所得出目標族群或次族群的療效反應者比例)
- the safety issues identified in the clinical investigation data and/or literature review and post-market data (clinical experience) for the intended purpose(s), as well as reasonably foreseeable hazards associated with the clinical use of the device that the data may not have captured e.g. misinterpretation or misuse of the device (其他尚未發現的安全問題或可預見的危害)
- the probability of patients experiencing a harmful event, that is, the proportion of the intended population that would be expected to experience a harmful event and whether an event occurs once or repeatedly may be factored into the measurement of probability (預期族群中會承受傷害事件的比例,也要考慮這些事件是單次發生或重複發生)
- the duration and severity of adverse events caused by the device or the procedure (由器材或程序所引發的不良事件的持續時間與嚴重度)
- whether there are mitigation strategies that have been implemented to address real or theoretical safety issues i.e. risk management documentation and IFU/labelling (是否已執行降低風險措施)
- any issues of uncertainty surrounding the application of the device for its intended purpose, e.g. limitations in the statistical analysis, generalisability of results to an Australian population (任何不確定問題)
| TGA 指引 (P.38) 還另外說,在此階段要考慮到「Product labelling」,要看「Labeling」與「Clinical data」是否一致,並確認「殘留風險 (Residual risks)」是否已充分揭露於使用說明書中。 也就是說,重點在於確認「Clinical evidence」、「Intended purpose」、「Indications」、「Contraindications」、「Warnings」、「Precautions」與「Actual and potential adverse effects」是否一致。 |
最後,結論要包含以下四點:(TGA 指引,P.38)
- Clinical evidence demonstrates compliance with EP 14 and the other EPs
- Clinical evidence on the device and/or substantially equivalent device is supportive of the safety and performance of the subject device
- Residual risks have been adequately mitigated with appropriate justification, for example, inclusion of relevant statements in the IFU and risk management documentation, and through post-market clinical follow up studies
- Risks associated with the use of the subject device are acceptable when weighed against the benefits to the patient.