筆記目錄
1. 前言
於日本境外設計、製造、且欲販賣醫療器材到日本境內的製造廠,須遵循於 2014 年 11 月 25 日生效的《PMD Act》(Pharmaceuticals and Medical Devices Act,藥品及醫療器材法),其前身為《JPAL》(Japanese Pharmaceutical Affair Law,日本藥事法)。
好學不倦的筆友,可至 Japanese Law Translation 搜尋《PMD Act》英日對照完整版詳閱。
(《PMD Act》的法規全名為《Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics》)
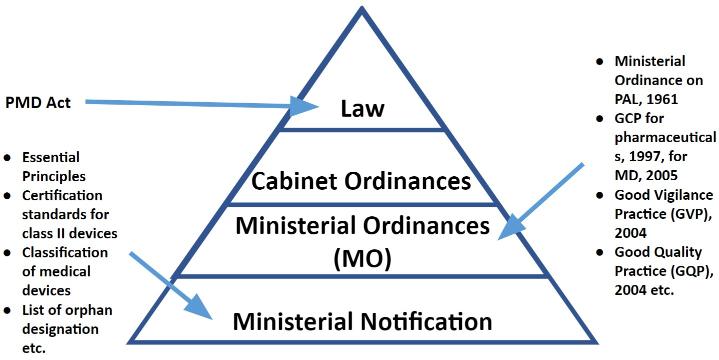
圖/改編於 Johner Institutr 的「Market Access of Medical Devices in Japan」,以及 PMDA 的「Understanding Japanese Medical Device Requirements」。
《PMD Act》由 MHLW (Japan’s Ministry of Health, Labour and Welfare,厚生勞動省) 負責,偕同 PMDA (Pharmaceuticals and Medical Devices Agency,醫藥品醫療機器綜合機構) 提供醫療器材、體外診斷 (IVD)、藥品、化妝品、可再生/細胞/基因治療等產品一個法規架構,涵蓋產品上市前申請/認證/審批、製造廠註冊、日本代表 (Designated Marketing Authorization Holder,DMAH)、品質管理系統、上市後安全監控…等等,以保障日本國人安全。
MHLW 就像是衛生福利部食品藥物管理署 (TFDA),負有政策與行政措施的最終責任;而 PMDA 類似財團法人醫藥品查驗中心 (CDE),是實際進行審查 (醫材上市前審查…)、檢查 (QMS/GLP/GCP 稽核…)、分析資料 (收集與分析不良事件報告…) 的單位。
分別於 2005 年 4 月與 2014 年 11 月,Third-party Registered Certification Body (RCB,算是日本的 Notified body) 開始可以進行 Class II 與 Class III Me-too 產品的上市審查,以確認產品是否符合上市標準。
另外,若有執行以下工作,位於日本境外的製造廠須在 PMDA 註冊工廠:
- Design and development
- Production or sterilization of medical devices
- Design and development or filling of in-vitro diagnostic reagents

圖/ QMS regulation in Japan (PMDA, issued on 1st April, 2015)
並且,海外製造廠須指定一個位於日本境內的 Designated Marketing Authorization Holder (DMAH),境外的註冊製造廠以及 DMAH 均須符合日本品質管理系統 (MHLW MO169)。
另外,若製造廠已取得其它地區的上市許可,例如美國、歐盟,則很多文件都可以沿用來申請日本上市許可。例如,日本的 Biocompatibility 及 Electrical safety 要求也是遵守 ISO 10993 和 IEC 60601;臨床試驗的要求則大多與 ICH E6 Good Clinical Practice (GCP) 相容。
但若欲申請上市產品和日本境內的類似品差異過大,或可能因人種差異導致安全功效的改變,還是其它區域的衛生照護環境、標準照護流程、醫材合併使用的狀況、病患組成 (Patient demographics) 與日本境內不同,此時就可能會被要求補做測試。
其實,要外銷醫療器材到日本,最首要的任務就是找到「日本代表」。有了日本代表,他們就會知道 (也必須知道) 境外製造廠要做什麼。
換句話說,如果不是個人興趣,那…Duck 不必往下看這篇筆記囉!趕快找到日本代表,依他們指示做事比較快。
2. 日本代表 (D)MAH
無論哪一等級均須指定一位日本代表,可分為 Marketing Authorization Holder (MAH: “Seihan” in Japanese) 和 Designated Marketing Authorization Holder (DMAH: “Sennin seihan” in Japanese) 兩種。
DMAH是海外製造廠所指定日本境內的代表,類似 Authorised representative (AR,歐體代表),所有的上市許可申請、上市後警戒、通報等都要透過 DMAH 執行。(IVD、Drug 和 Medical device 的 (D)MAH 不同,各自有不同的 Licenses)
| 雖然日本境外製造廠須指定 DMAH 始可申請產品上市,但製造廠可透過 Foreign Exceptional Approval System (外国特例承認制度),將產品註冊在自己名下,自己成為 Foreign Restrictive Authorization Holder (FRAH),而非 DMAH 名下。 |
| 依照 PMDA 的 FAQ,目前應該無法得知有哪些 (D)MAH: Q. Will PMDA provide a list of Japanese Marketing Authorization Holders? A. PMDA is NOT able to provide specific information such as a list of Marketing Authorization Holders. For your information, we only provide a link page including Japan’s pharmaceutical and medical device manufacture association which Japanese Marketing Authorization holders belong to at. – PMDA | Frequently Asked Questions (FAQ) | Q1-3 |
2.1 (D)MAH 的差異 & 組成
以下簡介 MAH 與 DMAH 的差異:
| 角色 | 區別 | 權限 | 如何選擇 |
|---|---|---|---|
| MAH | 日本境內已取得 Marketing authorization license 的製造廠。 | 是產品上市前認證/審批的申請者 (Applicant),也是產品上市許可的擁有者。 若有需要,可自由地轉換上市許可。 | 若是境外大型製造廠,可直接請日本境內的分公司作為 MAH 即可。 |
| DMAH | 由已取得產品上市許可的境外製造廠指定位於日本境內的 MAH 作為其 DMAH。 | 境外製造廠為上市前認證/審批的申請者,也是產品上市許可的擁有者。 DMAH 在申請上市認證/審批間 / 後,被境外製造廠授權代為執行相關作業。 須獲得境外製造廠的授權,DMAH 始可轉換產品上市許可。 | 若是中小型日本境外製造廠,找專職 DMAH 或 Distributor 作為 DMAH。 兩者各有千秋,若請 Distributor 作為 DMAH,好處是可能不用費用,但未來要更換 Distributor 時可能較為麻煩,且申請上市前認證/審批須繳交許多研發/製造機密文件,Distrubutor 相比於專職 DMAH 有較高的洩密機會。 |
2.2 (D)MAH 的組成
(D)MAH 由以下三種角色組成,各自在監督銷售、製造以及產品最終放行方面有不同的權責:
| 角色 (Controller) | 權責 | 擔任條件 |
|---|---|---|
| General Manager | 監督 (D)MAH | 1. 擁有工程或科學學士學歷,且 2. 在醫材或藥品公司擔任上市後安全 (Post-marketing safety) 或品保 (Quality assurance) 至少三年。 |
| Post Marketing Safety Controller | 1. 規劃與執行 Post-marketing surveillance (Good Vigilance Practice, GVP),以增進產品安全,且 2. 通知 PMDA 不良事件,並 3. 保留相關紀錄,必要時提供給 PMDA。 | 在相關領域至少有三年的 Good Vigilance Practice (GVP) 經驗 |
| Quality Assurance Controller | 1. 確保產品符合 QMS,包含:日本境內辦公室相關的 SOPs、倉儲、產品最終放行、召回、更新品保紀錄等;還有 2. 規劃產品的設計開發計畫並執行,包含 Verification/Validation。 | 至少有三年的品保經驗 |
| Pacific Bridge Medical「Local Agent Representation for Medical Products in Japan (DMAH)」整理不良事件通報期限如下: 1. 嚴重疾病與死亡 => 7 天內通報; 2. 身體其它疾病 (Physical disorders)、住院 (Hospitalization)、或其它非屬上一項的不良事件 => 14 天內通報; 3. 非屬上兩項的輕微不良事件 => 30 天內通報。 |
我時常被 GMP/GQP/QMS 搞混。參考下列 PMDA 2014 年的投影片「GMP and QMS Regulation in Japan」,可以協助釐清它們的差異:
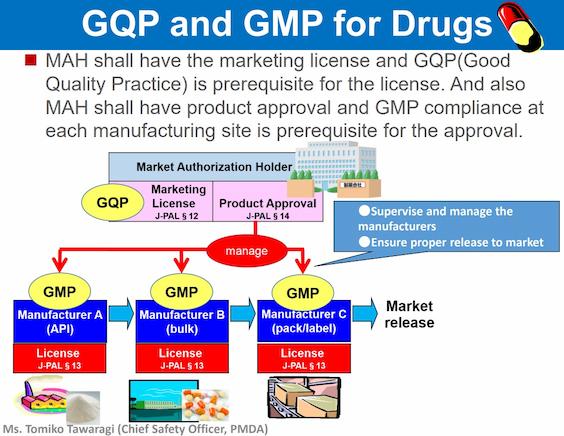

要當 (D)MAH,須先向 MHLW 申請執照 (KYOKA),且有不同等級的 (D)MAH:
| (D)MAH | 可負責的醫材等級 | 控制者 (Controller) |
|---|---|---|
| First class | 所有等級 (Class I ~ IV) | 至少須三人 (或兩人?) |
| Second class | Class I 及 Class II | 至少兩人才可組成 (D)MAH (Post Marketing Safety Controller 和 Quality Assurance Controller 須由不同人擔任,其中一人可兼任 General Manager。) |
| Third class | Class I | 一人即可組成 (D)MAH (一人可同時擔任三種 Controllers,承擔整個 (D)MAH 義務。) |
| Pacific Bridge Medical 在「Local Agent Representation for Medical Products in Japan (DMAH)」說:「Highly-Controlled Medical Devices (Class III and IV) must have three people for their MAH, and each individual acts as one Controller.」但, Johner-Institute 在「Market Access of Medical Devices in Japan」裡面寫道:「For a first class MAH, the general marketing supervisor must not be the safety manager. Therefore, at least two people are required for first and second class MAHs.」 所以,First class (D)MAH 至少要三人,或兩人即可? |
3. 分類分級
2005 年,日本以 Global Medical Device Nomenclature (GMDN) 為基礎,建立 Japanese Medical Device Nomenclature (JMDN) codes (Search database & List)。每一類醫材都有一個專屬的 JMDN code,每一個 Code 都有一個分類等級。
分為以下四級:
| GHTF Classification | PMDA Class | Category | Risk | Example |
|---|---|---|---|---|
| Class A | I | General MDs | Extremely low risk | Surgical instruments, including scalpels and tweezers, X-Ray film |
| Class B | II | Controlled MDs | Low risk | MRI scanners, digestive catheters, electronic endoscopes |
| Class C | III | Specially Controlled MDs | Medium risk | Artificial bones, dialyzer, mechanical ventilator |
| Class D | IV | Specially Controlled MDs | High risk (Invasive, potentially life-threatening) | Pacemaker, artificial heart valves, stents |
若沒找到適當的 JMDN code,可以依 Principles of Medical Devices Classification (GHTF/SG1/N77:2012) 來分級。
若 JMDN 第一碼為 1、3、或 4,代表是以 GMDN 為基礎所設立的;若第一碼為 7,則是 JMDN 獨有的代碼。例如:
- 40937 000 Nuclear medicine system workstation / Based on GMDN
- 70006 000 Arm type X-ray computed tomography system / Unique to JMDN
最後一碼代表風險等級 (Risk-based classification),若為 0,則代表此大類下沒有細分類。例如:
- 36957001 Body surface stimulator probe / Class Ⅰ
- 36957002 Subcutaneous stimulator probe / Class Ⅱ
- 36957003 Intramuscular stimulator probe / Class Ⅲ
- 36957004 Cardiac/central nerve stimulator / Class Ⅳ
最後兩碼表示此類的醫材風險等級都一樣,例如:
- 37626010 Mobile analogue general-purpose diagnostic X-ray system / Class Ⅱ
- 37626020 Mobile analogue general-purpose integral diagnostic X-ray system / Class Ⅱ
最後三碼則代表含有生物成分 (Biological ingredients) 或藥物 (Drug substances),例如:
- 10729100 Catheter for central venous
- 10729200 Catheter for antimicrobial central venous
- 10729300 Catheter for heparin-coated central venous
- 10729400 Urokinase-coated central venous catheter
QMS 證書效期 5 年,且須在到期前 6 個月開始更新。
4. 外國製造商註冊登記 (FMR)
無論等級的外國醫材製造商,所有負責設計開發 (Development) 、製造 (Production)、最終組裝 (Final assembly) 的地點,均須由 DMAH 向 MHLW 遞交 Form 63-5 申請 Foreign Manufacturer Registration (FMR),成為 Accredited Foreign Manufacturer,此流程稱為「Gaikoku seizo-gyosya nintei (外国製造業者認定)」。
FMR 證書效期為 5 年,MHLW 建議於到期日前 5 個月開始更新證書。
可以到 PMDA「外国製造業者認定・登録番号の公表」查詢目前已獲核准的外國製造商的相關資訊,例如,國別、廠址、有效年月日。

5. 品質管理系統 (MHLW MO169)
| 原本以為要先品質管理系統稽核後,再進行 FMR。但〈QMS regulation in Japan〉(PMDA, 2015) 寫:「Manufacturer needs “Registration” before the QMS inspection is conducted. 」 所以,FMR 應該比較像台灣的工廠登記。 |
所有等級的醫材製造商均須遵循 MHLW MO169 (Ordinance #169),此第169号法令是基於 ISO 13485 所建立的。在 2021 年 3 月 26 日 MO169 為了調和 ISO13485:2016 而改版,轉換期為 3 年,因此 DMAH 最遲須在 2024 年 3 月 25 日前符合此版本的 MO169。(版本差異請參考 PMDA「Revision of Japanese Medical Device QMS requirements」)
MO169 第三章節針對 (D)MAH 額外規定 QMS 文件/紀錄的保存期限、不良事件通報、優良警示規範 (Good Vigilance Practice,GVP) 等等。(細節請參考 PMDA「Tentative translation of MHLW MO 169 Chapter 3, as revised in 2021」)
大部分 Class II 醫材 (Specified Controlled) 的製造商品質管理系統 (QMS) 由 RCB 稽核;部分 Class II (Controlled) 以及 Class III、Class IV 製造商的 QMS 則須由 PMDA 稽核。
上述稽核均是在核發產品上市許可前稽核 DMAH 以及 Accredited Foreign Manufacturer,可能透過現場稽核或文件稽核,視是否有 ISO 13485、通報事件、召回,或依製程複雜度而定。
PMDA 一般對境外製造廠大多為文件稽核 (Document audit)。文件稽核時會確認下列資料:
- 製造現場總覽
- 組織架構
- 品質手冊
- 品質文件列表
- 製造流程 (含確效)
- MAH 合約
- 上市後警戒程序
若製造廠已有 Medical Device Single Audit Program (MDSAP) 稽核報告,一般來說,可免於重覆稽核的問題。

圖/ QMS regulation in Japan (PMDA, issued on 1st April, 2015)

圖/ QMS regulation in Japan (PMDA, issued on 1st April, 2015)
Class II 到 Class IV 醫材的 QMS 認證將由 PMDA 或 RCB 頒發。
| 合格的 RCB 名單可至 MHLW「登録認証機関制度について」查詢,例如 SGS、TÜV Süd、TÜV Rheinland、BSI 都是 RCB。 |
QMS 證書效期 5 年,且須在到期前 6 個月開始更新。
6. 上市前註冊/認證/審批 (Marketing approval,製造販売承認)
| PMDA 在 FAQ 裡說,所以上市申請的相關文件都須為日文: Q. Can we file our application for product approval to PMDA in a language other than Japanese? A. No, PMDA does not accept applications in other languages. Japan’s Pharmaceutical Affairs Law requires all forms related to the marketing application to be submitted in Japanese. – PMDA | Frequently Asked Questions (FAQ) | Q1-4 P.S. 申請書等文件與技術文件摘要 (STED) 須以日文填寫,但提交的測試數據報告,包括臨床前試驗與臨床試驗的報告,可以用英文撰寫。(Refer: 臺灣及日本醫療器材查驗登記常見問答集) |
6.1 各流程簡介
| 流程 | 流程名稱 | 審查單位 | 適用等級 | 上市途徑 | 解釋 |
|---|---|---|---|---|---|
| 上市前註冊 (Pre-Market Submission, PMS) | todokede 届出 | 自我宣告 | Class I | Self declaration | 1. DMAH 代表製造廠繳交相關文件到 PMDA。PMDA 不會進行實質審查,也不會給出任何意見。 2. 繳交文件大概有「製造廠資訊」,例如廠址、製造流程…,以及「產品基本描述」,例如名稱、預期用途、規格…。 |
| 上市前認證 (Pre-market certification, PMC) | Ninsho 認証 | Third-party Registered Certification Body (RCB) | 1. Class II (大部分有產品標準) 2. Class III (小部分有產品標準) | Third party Certification (Certification by a registered certification body is required.) | 1. 有相關標準 (JIS,很多是以 ISO/IEC 標準為基礎) 醫療器材。 2. DMAH 代表製造廠向 RCB 提交相關文件,並由 RCB 審查確認是否符合相關標準與 Essential principles。 3. 審查大約耗時 4 個月。(此為 Johner Institute「Market Access of Medical Devices in Japan」預估的時間,另可參考其它兩家顧問公司的經驗。) |
| 上市前審批 (Pre-Market Approval, PMA) | Shonin 承認 | PMDA 審查,MHLW 核准 | 1. Class II (小部分沒有產品標準) 2. Class III (沒有產品標準) 3. Class IV | Minister’s Approval (Reviewed by PMDA and approved by the MHLW) | 1. 目前大部分 SaMD 因為沒有產品標準 (Certification Criteria),所以都走此路徑。 2. 須符合 Review Guideline 3. 審查大約耗時 12 個月。(此為 Johner Institute「Market Access of Medical Devices in Japan」預估的時間,另可參考其它兩家顧問公司的經驗。) |
以上產品均須符合 Essential principles (MHLW Notification No.122 of 2005),且產品認證效期永久。
另外,執行 Pre-market certification (PMC) 的第三方認證單位須為 MHLW 核准的,名為「Registered certification bodies (RCB)」,也只有擁有 Certification criteria 的醫材可以被 RCB 認證而上市。
至於什麼是「Certification criteria」呢?PMDA 解釋如下:
“Certification criteria” mean the criteria for medical devices, for which compliance of medical devices is assessed by registered certification bodies that review applications of the devices. This criteria is specified by the Minister of Health, Labour and Welfare.
PMDA | About Criteria in Regulation | What is Certification Criteria?
那什麼是「Approval criteria」?
‘Approval criteria’ mean the criteria, compliance with which is assessed for regulatory approval of medical devices. ‘Approval criteria’, composed of ISO or IEC standards, etc., in principle, are intended for products for which no clinical data are required to be submitted.
PFSB Notification No. 1120-5, dated on November 20, 2014 (找不到 PMDA 在此引用的原始文件)
什麼是「Review Guideline」?PMDA 解釋如下:
The “review guidelines” are specified by MHLW and describe the major technical requirements and/or the acceptance criteria, etc. necessary to evaluate the safety and efficacy of medical devices, although no specific performance limits are determined for each technical requirement. The guidelines contribute to more efficient PMDA reviews of medical devices.
PMDA | About Criteria in Regulation | What is Review Guideline?
相關審查費用時常更新,因此需要時,可至 PMDA 官網搜尋「医療機器 審査手数料」,即可找到最新的費用列表。
6.2 備審文件
如同歐盟,日本的上市許可審查文件也須含 Clinical evaluation,以證明產品的安全與功效。一般 Generic/me-too 醫材的臨床評估報告以搜尋文獻找到的結果應已足夠,但若是 Novel 醫材,則通常須執行臨床試驗。而 Improved 醫材,視其資料完整程度而定,若無法用臨床前測試資料與文獻證明安全與功效,則要有臨床試驗。
| Generic/me-too MDs | Improved MDs | Novel MDs |
|---|---|---|
| Substantially equivalent | Non-substantially equivalent, but not novel。 可細分為以下四類: 1) with clinical data; 2) without approval criteria, without clinical data; 3) without approval criteria, without clinical data; 4) with approval criteria, without clinical data | New medical devices (Have a clearly different structure, usage, indication, performance, etc., as compared to those which have already been approved for marketing) |
所有備審文件都須是日文,且遵循 IMDRF 的 STED format。
| P.S. 申請書等文件與技術文件摘要 (STED) 須以日文填寫,但提交的測試數據報告,包括臨床前試驗與臨床試驗的報告,可以用英文撰寫。(Refer: 臺灣及日本醫療器材查驗登記常見問答集) |
6.3 如何獲得已上市產品列表
可於 PMDA「Reviews and Related Services | Medical Devices」看到經由上市前審批 (Pre-Market Approval) 路徑上市的產品資訊及審查報告,以及「Search system for each product type (in Japanese) | 医療機器 情報検索」可查到產品說明書。
7. 上市後階段 (Post-market)
7.1 上市後變更
以下兩份日本指引,依變更程度分成以下四種情況:
- MHLW Notification Yakushokukihatsu No.1023001 (October 23, 2008)
- Decision Tree for Determining Needed Procedure to Change Pre-market Approval (January 2010), issued by PMDA
| 變更類型 | 範例 | 注意事項 |
|---|---|---|
| New submission | 變更工作原理 (Operating principle) | 須符合日本品質管理系統 (MHLW MO169) |
| Partial change (IchibuHenkohShinsei) | 變更工作原理 (Operating principle)、材料、使用者介面。 | 須提出「Partial change application」,並符合日本品質管理系統 (MHLW MO169)。 |
| Minor change (KeibihenkoTodoke) | 新增不同尺寸,且此新增尺寸在 PMA/PMC 核准的範圍內。 例如,PMA/PMC 核准軸徑為 3mm、5mm、10mm,現新增 4mm 及 7mm 的軸徑。 | 變更後 30 天內通報,並符合日本品質管理系統 (MHLW MO169)。 |
| No regulatory procedure is needed | 非醫材主體的變更,且不影響醫材的安全與有效。 例如,變更包裝形式,從 5 個一包改成 10 個一包。 | 須符合日本品質管理系統 (MHLW MO169) |
另外,通常各國法規是基於風險考量這個普世通用的概念,來決定變更管制要做什麼 (變更許可證?廠內變更即可?) ,所以筆友也可參考以下兩篇相關筆記:
- 上市後醫材的設計 (工程) 變更多大會影響到許可證? 關於歐盟 CE MDR 的「過渡期」規定,本筆記整理給你!
- 上市後醫材設計 (工程) 變更會影響到許可證?關於美國 FDA、台灣食藥署 TFDA 的規定,本筆記整理給你!
7.2 上市後警戒 (Surveillance)
相關要求規定在 Good Vigilance Practice ordinance (Ministerial Ordinance 135)。
不過可以由 MDSAP Companion Document 第四章「Medical Device Adverse Events and Advisory Notices Reporting」快速了解日本的要求。簡而言之,無論發生在日本境內或境外,當發生嚴重不良事件時,製造廠都有義務立即通知 DMAH。(請參考 PMDA 的通報網站「医療機器の不具合等の報告方法(企業向け)」)
另外,也可由 PMDA 網站「不具合が疑われる症例報告に関する情報」查詢不良事件通報紀錄。
8. 整個流程需要多久
依照森維國際認證集團整理的資料,從一開始到產品開賣,需要八個月:
| 階段任務 | 時間 (月) |
|---|---|
| 1. 準備階段:確認產品分類分級,與選擇 DMAH | 1 |
| 2. 工廠登記 | 1 |
| 3. 品質管理系統審核 | 3 |
| 4. 上市前註冊申請/認證/審批 | 3 |
| 關於完成「品質管理系統審核」的所需時間,PMDA 在「Application for Accreditation of Foreign Manufacturers」說 (P.2): 「A target period to complete administrative processing (standard administrative process time) of accrediting a foreign manufacturer is not specifically set. However, the period can be estimated to be about 5 months because the Minister’s licensing process for a domestic manufacturing establishment takes about 5 months.」 |
另外,Freyr 顧問公司認為所需時間如下:
| Device Class | Approval Routes | Agency | Timelines (Month) |
|---|---|---|---|
| Class I | Pre-Market Submission (Todokede) | N/A | 1 |
| Class II | Pre-Market Certification (Ninsho) | Registered certification bodies (RCB) | 6 |
| Class II | Pre-Market Approvals (Shonin) | PMDA | 9 – 24 |
| Class III | Pre-Market Approvals (Shonin) | PMDA | 9 – 24 |
| Class IV | Pre-Market Approvals (Shonin) | PMDA | 9 – 24 |
in Japan
9. 參考資料
- UNDERSTANDING THE REGULATORY REQUIREMENTS OF THE JAPANESE MEDICAL DEVICE MARKET (TUV SUD)
- Authorization of Medical Devices in Japan (Johner Institute)
- Local Agent Representation for Medical Products in Japan (DMAH) (PACIFIC BRIDGE MEDICAL)
- Understanding Japanese Medical Device Requirements (PMDA)
- Criteria for Medical Devices (PMDA)
- 日本醫藥品 PMDA 認證 (森維國際認證集團)
- Medical Devices Regulatory Services in Japan (Freyr)
- GMP and QMS Regulation in Japan (PMDA)
- QMS regulation in Japan (PMDA)
- Application for Accreditation of Foreign Manufacturers (PMDA)
- Frequently Asked Questions (FAQ) (PMDA)
- Medical Device Change Applications in Japan (EMERGO)
- Regulations and Approval/Certification Process of Medical Devices (PMDA)