筆記目錄
一、前言
希望本文可幫助在美國和台灣販賣醫療器材的廠商去了解,上市後的設計變更 (aka 工程變更) 是否會影響到上述地區的銷售許可。換句話說,本文整理了美國與台灣官方單位的「變更是否達到申請變更的閾值 (Regulatory threshold)」的判斷依據。
以美國 FDA 來說
經由 510(k) 路徑上市的產品 (510(k)-cleared device),其後續變更是否會需要重新申請 510(k) 的參考依據,有以下兩本指引:
- 〈Deciding When to Submit a 510(k) for a Change to an Existing Device〉
- 〈510(k) for a Software Change to an Deciding When to Submit a Existing Device〉
兩者都是 2017 年 10 月 25 日發行,前者 (General modifications guidance) 涵蓋已取得 510(k) 核准醫材的所有變更評估,除了軟體相關變更的評估。
FDA 在本篇指引說到:
This guidance does not address software changes or modifications. Please refer to FDA’s guidance Deciding When to Submit a 510(k) for a Software Change to an Existing Device.
General modifications guidance | III. Scope | Software Changes
後者 (Software-specific modifications guidance),將專門說明軟體 (含 Firmware、SaMD) 變更多大才需要申請新的 510(k)。
換句話說,所有軟/韌體以外的變更評估,都必須參考〈General modifications guidance〉。下面是 FDA 的說明:
This guidance specifically addresses software modifications and is intended as a companion to Deciding When to Submit a 510(k) for a Change to an Existing Device. Any modifications that are not modifications to software are not within the scope of this guidance; such changes (e.g., labeling changes) should be evaluated using Deciding When to Submit a 510(k) for a Change to an Existing Device.
Deciding When to Submit a 510(k) for a Software Change to an Existing Device | III. Scope
上述兩本指引均不適用於 510(k)-exempt device 或 PMA device。
以台灣食藥署 (TFDA) 來說
〈有關醫療器材之標籤、仿單或外盒變更,無需申請核准之項目〉(衛署藥字第 89006412 號公告) 詳列核准仿單、標籤、包裝的哪些項目變更時,不需申請許可證變更。但這份公告距今 (2021 年) 已 21 年,當然是非常落伍的規定。
現代很多醫材都包含軟體,甚至軟體自己本身就是醫材 (SaMD),而查驗登記審查員又會要求廠商將送審時的軟體版本註明於核定中文仿單中。若廠商解讀此份公告為「不在這份公告內的變更,就是須要申請變更許可證」的話,依軟體三、五天就改版的頻率,廠商和 TFDA 勢必會被煩死、累死。
因此,CDE 審查員給我們建議:
廠商須自行在品質系統內建立變更評估流程,將軟體變更程度分級,並規定變更程度達多少 (閾值),須變更 TFDA 的許可證。
若廠內變更評估流程判斷某次軟體變更程度未達閾值,廠商可將相關評估文件留存備查,並自行更改中文仿單內註明的軟體版本 (e.g. V.1.0.0 改成 V.1.0.1),而不需申請變更許可證。有關上述的變更評估流程,目前 TFDA 沒有這類的指引,但可能今年 (2021 年) 會發布。在發布前,廠商可參考美國 FDA 相關指引。
因此,在看〈有關醫療器材之標籤、仿單或外盒變更,無需申請核准之項目〉時,比最好的解讀方式是:「若變更項目詳列於此公告中,則一定不須變更許可證;若變更項目不在這份公告內,則不一定要變更許可證。」另搭配 FDA 相關指引較為恰當。
| TFDA 於: 1. 2021 年 12 月公告〈醫療器材軟體上市後變更申請之管理指引〉(FDA器字第1101660098號公告); 2. 2022 年 3 月發布「醫療器材軟體上市後變更申請常見問答集」。 |
依附 《醫療器材管理法》 而一同生效的《醫療器材許可證核發與登錄及年度申報準則》第十四條,增列「無需申請核准之變更項目」如下:

同樣地《醫療器材許可證核發與登錄及年度申報準則》第十三條,也規範當有下列變更時,須向中央主管機關提出申請:
- 中文品名;
- 英文品名;
- 原廠標籤、說明書或包裝;
- 成分、材料、結構、規格或型號;
- 效能、用途或適應症;
- 製造業者名稱;
- 製造業者地址或製造國別;
- 許可證所有人;
- 許可證所有人名稱。
以歐盟 (MDR) 來說
請參考〈上市後醫材的設計 (工程) 變更多大會影響到許可證? 關於歐盟 CE MDR 的規定,本筆記整理給你!〉
二、基本概念
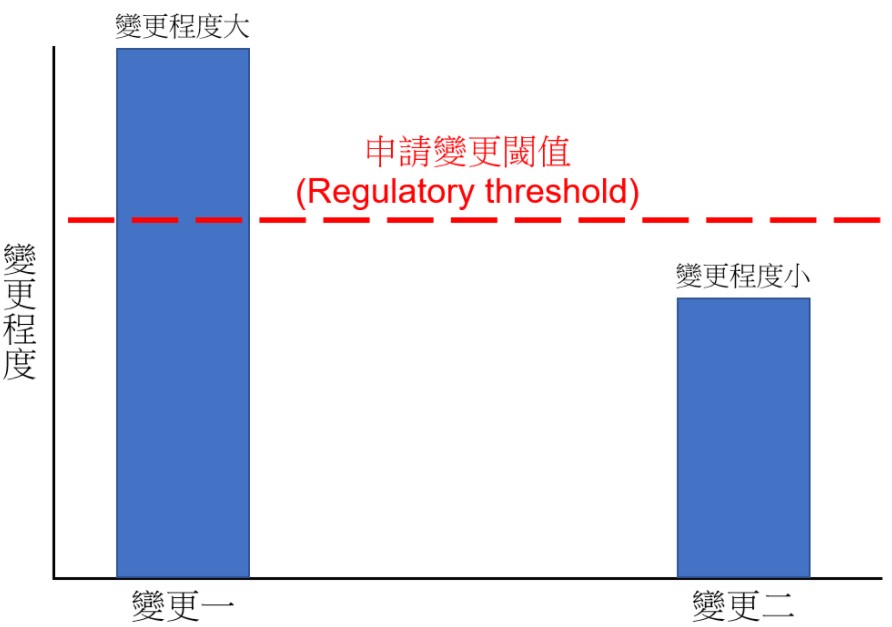
以下圖來說明,「變更一」超過申請變更閾值 (Regulatory threshold),代表變更對安全與功效的影響程度大到須申請新的許可證,或變更許可證。反之,「變更二」沒超過此閾值,只要依循廠內品質系統的變更管制 (21 CFR 820.30 (i)、820.70 (b)、IS0 13485:2016 Sec 7.3.9) 執行相關的驗證、確效、審查、核准,並將變更流程產生的紀錄好好留存備查即可。
以美國為例,這個閾值 (Regulatory threshold) 就是:
1) could significantly affect the safety or effectiveness of the device, or
2) major change or modification in the intended use of the device
21 CFR 807.81(a)(3)
廠商要評估變更是否超過閾值,決定是否要申請新 510(k) (原文如下):
when a change in a medical device would trigger the requirement that a manufacturer submit a new premarket notification (510(k)) to the Agency.
General modifications guidance | I. Introduction
總之,無論美國或台灣,我們要「了解 -> 評估 -> 決定」:
- 了解官方單位對於變更申請閾值 (Regulatory threshold) 的高低寬嚴,並且
- 每次變更時,評估是否有超過此閾值,
- 最後,決定變更後的動作:可能是 a) 變更許可證、申請新的 510(k)、或 b) 留存紀錄備查。
三、細說美國 FDA 的上市後申請變更閾值
關於軟/韌體以外的變更
背景介紹
在 21 CFR 807.81(a)(3) 規範了當下列重大變更 (Significantly changed or modified) 發生時,已上市醫材 (Currently has in commercial distribution or is reintroducing into commercial distribution) 須申請新的 510(k):
- 變更顯著影響到安全或功效 (Significantly affect the safety or effectiveness);
- 預期用途的重大變更 (Major change or modification in the intended use)。
看完上面的定義,應該大家都會想,「怎樣程度的變更是『Significant』,『Major』又是怎樣呢?」
換個方式問,「怎樣程度的變更才算超過申請變更閾值 (Regulatory threshold) 而須申請新的 510(k)」呢?
〈General modifications guidance〉透過「When to submit」決策流程協助我們確認 21 CFR 807.81(a)(3) 規範的閾值。
十大指導原則
在將來使用「When to submit」決策流程決定是否須要申請新的 510(k) 的過程中,要遵循以下十大指導原則,因此,請大家將原則熟記在心。

指導原則一、意圖重大變更者,通常要重申請 510(k) vs. 沒此意圖者,仍須評估是否超過閾值
| 變更類型 | 變更是否超過閾值 (是否需申請新的 510(k))? |
|---|---|
| 廠商意圖重大變更。 例如,想要顯著增進臨床成效、想降低某個已知的重大風險、或為了某件不良事件而進行的變更。 | 此類變更不須評估有無超過閾值,直接判定為「顯著影響安全與功效」,都須申請新的 510(k)。 (A change intended to significantly affect the safety or effectiveness of the device is considered to be a change that “could significantly affect the safety or effectiveness of the device” and thus requires submission of a new 510(k) regardless of the considerations outlined below.) |
| 廠商無意圖重大變更 | 就算廠商「感覺」這次變更很小,也「沒想要」重大變更什麼,但仍須依照本指引進行評估。 |
| 廠商為了處理某項違規或召回事件而進行的變更 | 參考〈Distinguishing Medical Device Recalls from Medical Device Enhancements〉決定後續動作。 |
指導原則二、著重 Safety & Effectiveness,而非僅 Harms 和其對 Safety 的影響
在接下來的「When to submit」決策流程中,須要注意變更有無
- 造成新的風險,或
- 是否有影響現有的風險。
FDA 在這邊強調,〈IS0 14971〉注重危害 (Harm) 和其對安全 (Safety) 的影響,但當評估變更是否超過閾值的過程中,廠商須注意的是安全 (Safety) 與功效 (Effectiveness)。
因為這些差異,FDA 特意用「Risk-based assessment」來提醒廠商在評估變更是否超過閾值時,須注意安全 (Safety) 與功效 (Effectiveness),而不使用「Risk analysis methods」,避免廠商忽略變更對功效的影響。
以下整理差異:
指導原則三、注意是否有非預期的影響
廠商在評估「計畫中的變更」是否顯著影響安全與功效後,也須注意此「計畫中的變更」是否引起其他「非預期的影響」,而後者也須被評估是否有超過閾值。
例如,變更滅菌方式可能會影響到產品材料,或變更產品材料可能會影響到產品功效。
指導原則四、納入風險管理概念
| 我將本篇指引的「E. Considerations for Risk-Based Assessments of Modified Devices」整理合併於此。 |
廠商須有〈ISO 14971〉的風險管理概念,例如,Hazards (危害)、Hazardous situations (危害情況)、Risk estimation (風險估算)、Risk acceptability (風險可接受度)、Risk control (風險控制)、Risk/benefit analysis (利益風險比分析)、Overall risk evaluation (總體 (殘餘) 風險評估)。
| 有關 Risk/benefit profile、Benefit-risk ratio、Benefit/risk determination 等等的利益與風險評估,請參考另篇筆記「醫療器材上市前審查考量之利益與風險權衡要素 (Benefit-Risk evaluation)」。 |
本篇指引引用〈ISO 14971〉對風險 (Risk) 的定義如下:
The concept of risk, is the combination of the probability of occurrence of harm and the severity of that harm.
(風險 (Risk) = 危害 (Harm) 發生機率 & 危害造成的嚴重程度)
若評估某次變更所導致的「危害 (Harm) 發生機率」低到可以忽略,則此變更應該不需申請新 510(k);若發生機率沒低到可忽略,又或者根本無法評估發生機率,則須進一步評估「危害造成的嚴重程度」決定是否達到申請變更閾值 (若嚴重程度也小到可忽略,應該也不需申請新 510(k))。
| 特別提醒:FDA 在此所指的「Risk」都是「Pre-mitigation risk (進行風險控制「前」的風險)」,不是降低風險後的風險喔。 |
但 FDA 還是特別提醒,本篇指引的風險管理概念與〈ISO 14971〉有些微不同,請見下表:
| 本指引的風險管理概念 | 〈ISO 14971〉的風險管理概念 |
|---|---|
| 使用〈ISO 14971〉風險管理概念時,常會著重在安全性,而忽略功效的評估。但廠商在這邊想 Risk 時,不只要考量安全 (Safety),還須考慮功效 (Effectiveness)。 在 21 CFR 807.81(a)(3)(i) 規定「A change or modification in the device that could significantly affect the safety or effectiveness of the device」時須申請新 510(k)。 因此,FDA 也提醒大家,變更時,除了考量安全,還須評估對產品功效的影響。 | 著重在產品的「安全性」 |
在評估變更影響時,若辨識出新風險 (New risks) 或顯著影響現有風險 (Significantly modified existing risks) 時,也就是超過「閾值」時,可能就須要申請新的 510(k)。
但,這邊指的「新風險」是:
1) 相較於變更前的產品 (Original device) 而言,變更後的產品被發現有新 Hazards (危害)、新Hazardous situations (危害情況) 是原本所沒有的 (a new risk is a new hazard or hazardous situation that did not exist for the original device),且
2) Pre-mitigation risk level (進行風險控制前的風險等級) 被判定為不可接受 (the pre-mitigation risk level associated with the new risk is not considered to be acceptable)。
General modifications guidance | 4. Use of risk management
另外關於「顯著影響現有風險」則是:
產品變更顯著影響了:
1) 風險評分 (Risk score,例如,RPN)、
2) 風險可接受類型 (Risk acceptability category),例如,原本「可接受」改成「不可接受」,反之亦然、或
3) 不利後果持續時間 (Duration,how long the adverse consequence lasts)。(a device change could be considered to significantly modify an existing risk if it changes the risk score, risk acceptability category, or duration of risk)
General modifications guidance | 4. Use of risk management
| FDA 在指引的 P.53 舉例如下: Change: A particular device heats fluid in order to achieve its intended effect. The most recently cleared device had a low-power heater and the maximum fluid temperature was low enough that the severity of the worst-case thermal injury was low to moderate. In the risk analysis for the design of the most recently cleared device, the risk score/rating for thermal injury was therefore in a range identified in the risk management document as “tolerable but undesirable,” before risk control measures were added. After receiving input from customers that the fluid heating process was too slow, the device was changed to use a higher-powered heater, which increased the maximum possible fluid temperature. Relevant questions: Q5 – Is it any other change in design (e.g., dimensions, performance specifications, wireless communication, components or accessories, or the patient/user interface)? Yes. Q5 – Does the change significantly affect the use of the device? No. This change would not significantly affect the use of the device. Q5 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? Yes. When the manufacturer performed a risk analysis on the new design, the severity of potential thermal injury increased and the risk of thermal injury became “unacceptable,” before application of additional risk control measures. This risk analysis showed that the design change had a potentially significant impact on safety by changing the pre-mitigation acceptability of the risk. Therefore, submission of a new 510(k) is likely required. This same conclusion holds whether or not the manufacturer needed to add new risk control measures to bring the final risk into the acceptable range. Decision: New 510(k). — 兩個重點: 1. 由此範例可了解,若因導入某變更改變而顯著修改風險評分,則須申請新 510(k)。 2. 這邊討論的風險、風險評分等,都是執行風險控制措施 (Risk control measure) 之前 (控制之後的風險如何,在評估是否達到申請新 510(k) 的閾值時是不管的。)。 (請參考後面「Q5. 有尺寸 (Dimensions)、性能 (Performance)、無線通訊 (Wireless communication)、材料 (Material)、零組件 / 附件 (Components / accessories)、病患或使用者介面 (Patient / user interface) 的變更嗎?」的相關筆記。) |
指導原則五、若 Verification & Validation 出現非預期結果,則可能須申請新的 510(k)
依據 21 CFR 820.30 的規定,無論是否要申請新的 510(k),變更後都須執行 Verification & Validation (V&V),以確認設計輸出符合設計輸入的要求 (Design output = Design input requirement)、以及產品符合使用者需求和預期用途。
FDA 在此段說:
If the initial decision following the risk-based assessment is that submission of a new 510(k) is not required, this decision should be confirmed by successful, routine verification and validation activities.
If routine verification and validation activities produce any unexpected results, any prior decision that submission of a new 510(k) is not required should be reconsidered.
General modifications guidance | IV. Guiding Principles
整理上述原文成表格如下:
| x | 情境一 | 情境二 | 情境三 |
|---|---|---|---|
| 先 Risk-based assessment 的評估結果 | 不須申請新的 510(k) | 須申請新的 510(k) | 不須申請新的 510(k) |
| 後 Verification & Validation (V&V) 結果 | 出現非預期結果 (或不足以驗證 / 確效安全與功效) | 沒出現非預期結果 | 沒出現非預期結果 |
| 是否須申請新的 510(k)? | Yes | Yes | No |
指引中特別說明,針對變更的 V&V activities 須是「Rutine」的,以下摘要此部分解釋:
“Routine” activities in this context refer to the original design verification and validation activities that were done to assess the original device design.
General modifications guidance | IV. Guiding Principles
指導原則六、須考量同時的個別變更,也須將這些變更綜合評估
同一時間可能進行很多變更,所以,除了個別評估每個變更外,也須綜合考量同一時間的所有變更。
指導原則七、多次變更累加後,可能會超過閾值
每次單一變更可能都不超過閾值,所以評估為不須申請新的 510(k)。但 FDA 也提醒廠商,要注意變更的累加效應 (Cumulative effect of individual changes)。
廠商須比較「變更的產品」和「原始產品 (Original device)」之間的差異,因為可能多次些微的變更,使此產品漸漸地與當初的原始產品差異很大 (類似「Predicate creep」)。
在這邊提到的「原始產品 (Original device)」,是指廠商
- 最近拿到 510(k) 核准的產品 (Previously found to be substantially equivalent in their most recently cleared 510(k))、
- 1976 年前已上市的產品 (Preamendment Devices)、或
- 經由 De Novo 程序上市的產品 (Device that was granted marketing authorization via the De Novo classification process)。
以下摘錄指引中「原始產品 (Original device)」的定義:
Original device: the device described in their most recently cleared 510(k) for the device, their legally marketed preamendments device, or their device that was granted marketing authorization via the De Novo classification process.
General modifications guidance | IV. Guiding Principles
指導原則八、建立變更評估紀錄程序 & 將每次變更的評估過程文件化
當執行變更時,廠商須符合美國 QSR (21 CFR Part 820) 的要求,除非產品不歸 QSR 管。Part 820 有很多要求,其中,有要求產品變更須被文件化。
因此,FDA 在此指引中說,廠商須在自己的品質系統內制訂變更評估流程的文件化程序,並且依此程序記錄每次變更。
指導原則九、若因變更須申請新 510(k),則送審文件中須描述其它次要變更
若變更導致須申請新的 510(k),則 510(k) 送審文件中,當然須描述所有修改程度到達閾值的變更內容。
要特別注意的是,若前次 510(k) 申請案的產品有 A、B、C、D、E 五個規格,而送審文件中只有描述到 A、B、C 三個規格。後續此五規格都有變更,但僅有 A 變更評估為須申請新 510(k),B、C、D、E 變更評估不須申請新的 510(k)。這種情況下,新 510(k) 申請案中要列出 A、B、C 三個變更內容,但不用列出 D、E。
FDA 舉例說:
510(k)s typically include a listing of device warnings in the labeling, so if a warning in the device’s labeling had been changed, that change should be described in the new 510(k), even if that change did not itself trigger the requirement for submission of a new 510(k).
However, a 510(k) would not typically identify or describe individual components of a circuit board, such as resistors, and therefore FDA would not expect changes to the resistors to be listed in the new 510(k) for a modified device because the first 510(k) would not have included information about the resistors.
General modifications guidance | IV. Guiding Principles
另外,若同時有多個變更,其中有些變更程度較大達到申請新 510(k) 的閾值,有些沒有。則那些未達閾值的變更不需等新的 510(k) 許可,可立即執行。
指導原則十、實質相等性 (Substantial equivalence) 評估
當變更程度大到超過閾值而去申請新的 510(k) 後,廠商仍須如同一般 510(k) 申請案般評估實質相等性 (Substantial equivalence, SE)。各位可參考本網站的〈如何使用 Multiple Predicates 來證明 Substantial Equivalence〉與〈如何用 Reference Devices 證明 510(k) 的實質相等性 (SE)?〉進一步深入了解 SE。
正式進入評估流程
FDA 將評估流程畫成一張主表、四張次表。四張次表分別用來評估:
- Labeling changes (次表 A)
- Technology, engineering, and performance changes (次表 B)
- Materials changes (次表 C)
- Technology, engineering, performance, and materials changes for in vitro diagnostic devices (IVDs) (次表 D)

主表 – 是否意圖重大變更?
此次變更意圖顯著增進安全 (Safety) 或功效 (Effectiveness) 嗎?
例如,1) 顯著增進臨床成效 (Significantly improve clinical outcomes)、2) 想降低某個已知的重大風險 (Mitigate a known risk)、或 3) 為了某件不良事件 (In response to adverse events) 而進行的變更,均為意圖顯著增進安全或功效。
若是,則可能這次變更程度已達閾值,須申請新 510(k)。
若否,則:
下列兩個例子說明「一個變更影響多個因素」時的評估方式:
範例一:變更製程
廠商想要將加工方式改為沖壓 (Stamping process),這樣的變動須修改不鏽鋼的等級,以及此物件的尺寸允收誤差。此時,須遵循次表 B及次表 C評估。(IVDs 則須看次表 D)
範例二:變更 Shelf-life
廠商變更一或多個材料以改善產品的 Shelf-life。這項變更還會影響到 Performance characteristics,也須更新 Labeling。此時,須遵循次表 A、次表 B及次表 C評估是否達到閾值。(IVDs 則須看次表 D)
若變更的類型不在次表 A ~D,該怎麼辦?
此時廠商須參考〈General modifications guidance〉的 Section E (Considerations for Risk-Based Assessments of Modified Devices) 進行評估。(請參考本文「指導原則四 (納入風險管理概念)」)
次表 A – Labeling Changes (九個評估重點)

[Q1. 是否修改 Indication for use?]
一般只要修改到 Indication for use 就會挑起 FDA 的敏感神經,因為可能稍微修改,就顯著影響產品的安全與功效。
這部分,大致可分為以下二類:
| 修改內容 / 目的 | 是否需申請新 510(k)? |
|---|---|
| 僅為了使語意更清楚,並不會影響涵義 (Meaning) 或其組成內容 (Substance) | No |
| 減 Indicaiton for use 的範圍 (Limit use within the currently cleared indication) (例如,原 510(k) 許可三個適應症,但此次變更限縮為二個) | No |
| 〈General modifications guidance〉雖然規定,若變更 Indication for use 則依 Q2 ~ Q6 評估;其餘部分的 Labeling Changes,則由 Q7 開始評估。 但是,有以下的敘述: “It should be noted the decision points in A1.1 (本文 Q2) - A1.5 (本文 Q6) may apply not only to changes to the indications for use statement of the labeling, but also to changes to other sections of the labeling, such as the directions for use of the device. (Page 17) Not all changes that describe a new disease, condition, or patient population that the device will diagnose, treat, prevent, cure or mitigate are necessarily made in the indications for use section of the labeling. These types of changes could also result from a change to other sections of the labeling, such as the directions for use of the device. (Page 18)“ 因為上述要求「修改 Indication for use 以外的地方,也須考慮是否有 Q2 ~ Q6 的可能」,所以,本文修改這部分評估流程。(請參考本指引 Page 16 的 Figure 2 – Flowchart A: Labeling Changes 以了解 FDA 原始流程) |
[Q2. 是否將一次性使用 (Single-use only) 修改為可重複使用 (Reusable)?]
Single-use only -> Reusable,通常須申請新 510(k);
Reusable -> Single-use only,則不需申請新 510(k)。
[Q3. 是否修改處方用 (Prescription, Rx)) 成為非處方用 (Over The Counter, OTC)?]
醫療照護專業人士 (Health care professionals) 與業餘使用者 (Lay users) 所需的使用指示 (Directions for use) 不同,通常因為後者沒有專業知識,對於使用指示的易讀性要求較高,所以:
Rx -> OTC,通常須申請新 510(k);
OTC -> Rx,則不需申請新 510(k)。
[Q4. 是否僅修改品名,或此修改僅為改善易讀性、使語意更清楚明瞭?]
若只是修改品名,或只是改善可讀性 (Readability)、使文義更加清楚,即使修改到原核可的 Indication for use,也不需申請新 510(k)。(須注意:修改後的 Indication for use 文義,不可與原核可文義有落差。)
[Q5. 是否新增新疾病、使用情境 (Condition)、病人族群 (Patient population)?]
評估變更 Labeling 是否新增新疾病、新使用情境、或病人族群,若有,表示此變更將顯著影響產品的安全與功效,因此須申請新 510(k)。
廠商可由下列五方面來評估:
- Patient demographics
- Diagnosis
- Prognosis
- Comorbidity (共病症)
- Complications (併發症)
若變更 Labeling 有明顯影響到上面五點,則須申請新 510(k)。
要注意的是,有時上述三要點 (疾病、使用情境、病人族群) 會被「間接」影響而導致須申請新的 510(k)。
例如,只是將使用指示由「有症狀時使用」改為「每天使用,即使無症狀」,這樣的修改可能導致此產品由「治療」變更為「預防」,而病人族群將由「有症狀、或被確診的人」變更為「無症狀、或健康的人」,如此則須申請新 510(k)。
[Q6. 以 Risk-based assessment 評估是否有新風險或顯著影響現有風險?]
若 Q1 到 Q5 都判定變更沒有到達申請新 510(k) 的閾值,廠商須依「指導原則四 (納入風險管理概念)」 進行 Risk-based assessment。變更 Labeling 可能會影響如何 (How) / 何時 (When) / 何處 (Where) / 誰 (By whom) 使用此產品,進而影響風險概況 (Risk profile)。
FDA 列舉五類變更,解釋如何以 Risk-based assessment 評估是否達到須申請新 510(k) 的閾值。
| 變更類型 | 討論 |
|---|---|
| anges to the type of joint, organ, bone, vasculature, or tissue applied to or interacted with, regardless of the section of labeling in which this information is contained | 變更產品使用 / 交互作用的部位 (Joint, organ, bone, vasculature, or tissue) 可能會導致 Q5 所說的「新疾病、新使用情境、或病人族群」,進而須申請新 510(k),以下列舉兩例: [範例一] 有一骨頭固定系統 (Bone fixation system,例:骨釘、骨板) 原本使用於四肢 (Extremity),變更為使用於頭骨 (Skull),則此種變更可能因顯著影響產品的風險概況 (Risk profile) 而須申請新 510(k)。 [範例二] 原使用於 A 長骨 (Long bone) 的骨頭固定系統變更為可使用於不同類型的 B 長骨,此類變更雖然也會有「新疾病、新使用情境、或病人族群」,但可能因沒顯著影響風險概況,故無需申請新 510(k)。 |
| Changes in user or use environment | [範例一] 使用環境由手術室變更為恢復室 (Hospital recovery room),兩者均有專業醫療照護,可能因此不會顯著影響產品的風險概況 (Risk profile)。 [範例二] 當一般醫療器材 (Basic medical equipment) 的使用者,由熟悉使用方法的家醫科醫師 (General physician) 變更為同樣熟悉使用方法的技師 (Specialist),可能因此不會顯著影響產品的風險概況,不需申請新 510(k)。 [範例三] 從專業用途 (Professional use) 變更為家用 (Home use),或由限於醫院使用 (Hospital use) 改成運輸中使用 (Ambulatory transport),這類變更很可能須申請新 510(k)。 因為從醫療專業使用情境改為家用情境,這樣的變更將導致使用情境由一個有專業醫療照護監督者在場的情況,變成可能僅有無醫療專業知識業餘者在現場的情況。 而且,家庭環境可能會有家用電器的電磁波影響 (EMC),或者潔淨度、電壓 / 電流不穩等因素導致安全或功效問題。 另外,原本在醫院使用,改為可在運輸中使用,也將導入許多變因,增加原本沒有的風險,例如,震動導致產品失效。 上述種種問題,可能將顯著影響風險概況。 |
| Changes in frequency or duration of use | 這類變更可能增加 / 減少產品的使用頻率、使用時間,或變更定期監控 / 長期連續監控。 |
| Changes concerning the compatibility or interoperability of a device with other devices, components, or accessories | 這類變更例如:1) 變更 IVD 試劑搭配的系統;2) Infusion pump 變更為可由脈搏血氧飽和儀 (Pulse oximeter)、血壓計輸入資訊 (原先不可由此二儀器輸入資訊)。 廠商須確認以下重點: [重點一] 變更前 / 後所搭配產品的差異 廠商須鑑別與評估產品相接的設備 (Device)、組件 (Component)、配件 (Accessory) 變更前 / 後的差異,例如,這樣的變更是否會影響生物相容性、性能 (Performance)、連接性 (Connectivity)。 若變更後所搭配的產品與原本搭配產品類型不同,則可能須要申請新的 510(k)。 [重點二] 關鍵性 (Criticality) 搭配的設備、組件、配件在整個系統中扮演越關鍵的角色,則這類變更越可能須申請 510(k)。 [重點三] 相接設備、組件、配件是否宣稱可與本產品搭配使用 若所搭配的設備、組件、配件的 Labeling 中已宣稱可與本產品搭配使用,這種情況下較不會有 Compatibility、Interoperability 的問題。 (PS.) IVD 產品請參考〈Replacement Reagent and Instrument Family Policy〉 |
| Changes from a general use to a more specific use | FDA 說這類變更是最難評估的,須參考〈General/Specific Intended Use〉。 |
| FDA 在指引的 P.46 舉例如下: Change: The directions for use of a catheter guidewire are modified to provide instructions on how to access different types of vasculature that were not previously addressed in the labeling. Relevant questions: Q9 – Could the change affect the directions for use of the device? Yes. Q5 – Does the change describe a new disease, condition, or patient population that the device is intended for use in diagnosing, treating, preventing, curing or mitigating? No. The guidewire is intended for use in the treatment of similar patient populations with the same diseases, even if the access points differ. Q6 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? Yes. The revised instructions suggest that the device can be used in new vasculature, which would be considered an expansion of the device’s indications for use. A risk-based assessment identifies that the new vasculature is more tortuous and significantly increases the risk of several device failure modes, which could significantly affect safety and effectiveness. Decision: New 510(k). — 特別摘錄這個範例是因為: 1. 在我看到「Modified to provide instructions on how to access different types of vasculature that were not previously addressed in the labeling」時,我直覺這會造成「New disease, condition, or patient population」,但 FDA 判定為「No」。 2. 在「Risk-based assessment」時,FDA 說這類的變更「Would be considered an expansion of the device’s indications for use」,呼應了前面提到的「雖然不是直接修改 Indication for use,但 Indication for use 有可能被其他變更影響」 |
[Q7. 是否新增 / 刪除 / 修改禁忌症 (Contraindication)?]
變更禁忌症通常會顯著影響產品的安全與功效,故大多這類變更的程度會大到超過須申請 510(k) 的閾值,但深入了解,可將「變更禁忌症」分成以下三類情況:
新增禁忌症
若廠商因收集到重要上市後資訊,須盡速新增禁忌症以保障社會大眾健康安全,FDA 並不反對廠商在申請新 510(k) 之前就新增禁忌症到 Labeling 上,並通知現有使用者。
此類因緊急情況而「先斬後奏」的 510(k) 稱為「Change Being Effected (CBE)」,與一般「510(k) 許可變更後才可修改」(先奏才能斬) 的方式不同。
但廠商修改完 Labeling 後,仍須申請新的 510(k),且在申請案的送審文件中應明顯標示此案是「Change Being Effected (CBE)」。CBE 510(k) 的概念應該是來自 FDA 對藥品的管制,以下節錄網友對 CBE 510(k) 的解釋:
For each significant change you need to submit a 510(k) Application before you implement this change to your device/system/labeling. Once the FDA approves this change, you can modify your device / labeling. It is different for contraindication.
CBE refers to the basic 510(k) application only that prior to 510(k) submission you can state this new indication to your official labeling for the US.
The regular fee for 510(k) has to be paid. The content of CBE 510(k) should be the same as regular 510(k). You are allowed to mark chapters that are the same as in previous 510(k) submission.
CBE (Change Being Effected) 510(k) Submission (elsmar.com) | med_cert
刪除禁忌症
通常須申請新 510(k)。
例如,身體約束 (Physical restraint) 產品原本禁止使用於體重 100 磅以下的人,若刪除此限制,則 Indication for use 會大幅擴張 ,將顯著影響產品的安全與功效。
修改禁忌症
同樣地,如果修改禁忌症會擴張 Indication for use,須申請新 510(k)。
但若只是 (透過 Clarify 或 Reword) 增進禁忌症描述的可讀性,不會變更到禁忌症所限制的內涵,則不需申請新 510(k)。
[Q8. 是否修改警語 (Warnings) 或注意事項 (Precautions)?]
廠商須於監控產品上市後相關的安全資訊,通常這類資訊會依據 21 CFR Part 803 (Medical Device Reporting regulation,MDR) 被通報至 FDA 資料庫。
若因為類似品的不良事件,廠商想防患未然將相關警語、注意事項新增 / 修改到 Labeling 中,FDA 認為此類變更大多不需申請新 510(k)。
但須評估完 Q9,才可確認是否未達申請變更閾值。
[Q9. 變更是否會影響到產品的使用指示 (Directions for use)?]
若變更不影響到使用方式 (How the device is used in practice),應該就不用申請新 510(k)。
若有影響,則須依循 Q2 到 Q6 評估變更幅度是否達到申請新 510(k) 的閾值。
以下列出 FDA 認為有影響使用指示的變更:
- Adding additional or new instructions on how to interpret diagnostic data from a diagnostic device.
- Adding a new procedural technique not described in the original labeling.
- Use of a product for a duration/frequency that is different than what is described in the labeling of the cleared device.
- Changing from labeling a device as non-sterile to labeling it as sterile or vice versa.
- Adding instructions for device use in a new patient population not described in the original indications for use.
- Adding instructions for device use in a different type of joint, organ, bone, vasculature, or tissue.
| FDA 在指引的 P.46 舉例如下: Change: The manufacturer adds a foreign language translation of the directions for use to a device’s labeling. The translation does not change the meaning of the instructions. Relevant questions: Q9 – Could the change affect the directions for use of the device? Yes. Q6 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? No. As long as the translation does not change the meaning of the instructions, this change would not affect the device’s risk profile. Decision: Documentation. — 特別有趣的是,「將操作指引翻譯成新的語言,但沒修改任何內容」屬於「會影響到產品的使用指示 (Directions for use)」,故我將此類變更收錄於此筆記。 |
| FDA 在指引的 P.47 舉例如下: Change: A manufacturer changes the design of an IVD for diagnosing herpes simplex 1 and 2 to a less strict performance specification that decreases both the sensitivity and specificity of the device to increase production. The manufacturer updates the performance specifications found in the labeling of the device. Relevant questions: Q9 – Could the change affect the directions for use of the device? Yes. The change could affect the directions for use by adding new instructions on how to interpret diagnostic data from the device. Q6 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? Yes. The changes to the device result in significantly increased existing risks. This is due to a mathematically expected increase in false positive results, which would, in turn, be expected to lead to an increase in harms such as mental anguish, delayed diagnosis for the true cause of any symptoms, and unnecessary treatment (e.g., pregnant women and newborns receiving unnecessary antiviral drugs or an unnecessary caesarean delivery of the fetus). Further, this would also significantly increase risks due to a mathematically expected increase in false negative results, which would, in turn, be expected to lead to an increase in harms such as delayed diagnosis that would in turn delay treatment of the underlying condition and could lead to unintended spread of the disease (e.g., through sexual partners, neonatal transmission during vaginal delivery, and transplanted organs). (未節錄 IVD 相關討論) Decision: New 510(k). — 摘要此範例原因如下: 1. 降低敏感度和特異性會影響「How to interpret diagnostic data」,進而修改「使用指示 (Directions for use)」。(請參考前面所列出六種可能影響「使用指示」的情況。) 2. 敏感度、特異性是原本就有的風險,現在降低標準,導致「顯著影響現有風險」。 |
| FDA 在指引的 P.48 舉例如下: Change: The manufacturer changes the material of the immediate container for an IVD reagent such that the shelf-life of the reagent is extended 3 months. As a result of the change in materials to the immediate container for the IVD, the labeling is updated to reflect the extended shelf-life. Relevant questions: Q9 – Could the change affect the directions for use? Yes. The labeling change to update the shelf-life could affect the instructions and directions for using the device. (未節錄 IVD 相關討論) Decision: New 510(k). — 由此範例可知,修改 Labeling 上的 Shelf-life 會影響「How the device is used in practice」。 |
次表 B – Technology, Engineering, and Performance Changes (五個評估重點)

[Q1. 是否為 IVD (In vitro diagnostic) 產品?]
若是,請跳過此部分,直接參考本指引的 Section D – Technology, Engineering, Performance, and Materials Changes for In Vitro Diagnostic Devices」。
[Q2. 是否變更控制機制 (Control mechanism)、工作原理 (Operating principle) 或能量形式 (Energy type)?]
重點一、變更控制機制 (Control mechanism)
例如,由機電控制方式 (Electromechanical control) 改成微處理器控制 (Microprocessor control)、由類比 (Analog) 控制改為數位 (Digital) 控制、或將呼吸照護機 (Respiratory care device) 由氣動控制 (Pneumatic control) 修改為電子控制 (Electronic control),都算變更控制機制。
幾乎這類變更的影響幅度都很大,大多會顯著影響產品安全與功效,因此須申請新 510(k)。
重點二、變更工作原理 (Operating principle)
同樣地,通常這類變更須申請新 510(k)。
例如,在醫療影像儲傳系統 (Picture archiving and communication system,PACS) 使用新的演算法去壓縮影像,或 IVD 產品由免疫螢光法 (Immunofluorescence) 改成 ELISA,都算是變更工作原理。
FDA 舉例,若將電腦斷層掃描 (Computed tomography,CT) 內的影像重建演算法 (Image reconstruction algorithm) 由單純的反投影法 (Back projection) 變更為新的演算法,這種變更幅度很大,除了實驗室的測試 (Bench test) 外,甚至還需透過臨床試驗確效。
或是,呼吸器用氣體潮濕器 (Respiratory gas humidifier) 的水滴散佈方式 (Water droplet dispersal method) 由壓電材料 (Piezoelectric Material) 改為 Wick and fan method,也會大幅影響安全與功效。
重點三、變更能量形式 (Energy type)
和上兩個變更一樣,這類變更大多須申請新 510(k)。
例如,輸入 (Input) 產品的能量形式由交流電 (AC) 改為電池,或將產品輸出 (Output) 的能量由游離輻射 (ionizing radiation) 變成超音波 (ultrasound),都屬於這類變更。
在此討論的「變更能量形式」不包含以下兩類:(此類變更請參考 Q5)
- 改變電壓,例如,3V -> 9V;
- 改變電池種類,例如,鎳鎘電池 (Nickel-cadmium battery,NiCd) -> 鉛酸電池 (Lead-acid storage battery)。
[Q3. 是否改變滅菌 (Sterilization)、清潔 (Cleaning)、消毒 (Disinfection)?]
有四個評估重點。
重點一、是否由「Established category A」滅菌方式改為「Established category B」或「Novel」的滅菌方式?
若是 (A -> B or Novel),則通常須申請新 510(k);反之 (B or Novel -> A),通常不需要。以下說明滅菌方式的分類 (詳細解釋請參考〈Sterility Information Guidance〉):
| Established category A | Established category B | Novel |
|---|---|---|
| 成熟方法,已有 FDA 認可的標準。 | 還沒 FDA 認可標準,但已有開發、確效等資訊可供 FDA 評估。 | 新穎方法,沒有足夠文獻證明其效能,FDA 也還沒確認此種滅菌方式可有效滅菌。 |
| These are methods that have a long history of safe and effective use as demonstrated through multiple sources of information such as ample literature, clearances of 510(k)s or approvals of premarket approval (PMA) applications, and satisfactory QS inspections. For established methods such as dry heat, EO, steam, and radiation, there are voluntary consensus standards for development, validation, and routine control that are recognized by FDA. | There are other established methods for which there are no FDA-recognized dedicated consensus standards, but for which published information on development, validation, and routine control is available. | These are newly developed methods for which there exists little or no published information, no history of comprehensive FDA evaluation of sterilization development and validation data through an FDA-cleared 510(k) or approved PMA for devices sterilized with such methods, and no FDA-recognized dedicated consensus standards on development, validation, and routine control. A Novel Sterilization Method is a method that FDA has not reviewed and determined to be adequate to effectively sterilize the device for its intended use. |
| 1. Dry heat 2. EO (in a fixed, rigid chamber) 3. Moist heat or steam 4. Radiation (e.g., gamma, electron beam) | 1. Hydrogen peroxide (H2O2) 2. Ozone (O3) 3. Flexible bag systems (e.g., EO in a flexible bag system, diffusion method, injection method) | 1. Vaporized peracetic acid 2. High intensity light or pulse light 3. Microwave radiation 4. Sound waves 5. Ultraviolet light |
重點二、是否降低滅菌保證水準 (Sterility Assurance Level,SAL)?
除非降低後的 SAL 仍優於 10-6,否則降低 SAL 通常須申請新 510(k)。
重點三、變更再處理產品 (Reprocessed devices) 的清潔、消毒程序,是否會影響後續處理?
再處理產品於使用後,可能會先經過前處理 (例如,清潔、消毒),再進行最終處理程序 (例如,滅菌)。若廠商變更了前處理程序,可能提高產品的生菌數 (Bioburden levels),導致最終處理程序失效,進而嚴重影響安全與功效。〈Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling〉的 Appendix E 列出相關高風險醫材,FDA 認為此附錄中的醫材,若沒有足夠的再處理程序,將有很高風險會傳染疾病。
重點四、變更產品提供給使用者時的滅菌狀態?使用形式是多人、單人多次、單人單次使用?
以「滅菌狀態」來說,可能有以下幾種變更:
- 出廠時有滅菌 -> 出廠時沒滅菌 (改由使用者自行滅菌,或產品不須滅菌即可使用)
- 出廠時沒滅菌 -> 出廠時有滅菌
上面兩種變更,都很可能須申請新 510(k)。
以「使用形式」來說,有以下幾種:
- Single-patient, single-use
- Single-patient, multi-use
- Multi-patient, multi-use
若由 1 -> 2、1 -> 3、2 -> 3,則很可能須申請新的 510(k);反之,若由 3 -> 2、3 -> 1、2 -> 1,因這樣的變更不太會顯著影響安全與功效,故應該不影響原有的 510(k)。
重點五、此變更是否會顯著影響產品性能 (Performance) 或生物相容性 (Biocompatibility)?
滅菌 (Sterilization)、清潔 (Cleaning)、消毒 (Disinfection) 變更時,須對以下兩方面進行評估:
| 產品性能 (Performance) | 生物相容性 (Biocompatibility) |
|---|---|
| 尤其要特別注意產品的高分子材料 (Polymeric materials)、表面塗層 (Surface coatings)、吸收材質 (Resorbable materials)、動物來原材料 (Animal-derived materials) 是否因變更而有所影響。 | 注意是否增加不適當物質的殘留量,或影響產品特性。 |
| 評估是否: 1. 改變產品材料的結構、 2. 導致新的風險、或 3. 顯著影響現有風險 (Significantly modified existing risks) (參考「指導原則四 (納入風險管理概念)」)、 4. Verification & Validation 出現非預期結果 (參考「重點四 (V&V 是否發現任何非預期的安全與功效問題)」) | 舉例如下: 1. 變更 EO 滅菌 (Ethylene oxide sterilization) 程序可能增加 EO 殘留量; 2. 變更清潔程序,可能會在與病患接觸的部位殘留不適當的清潔劑; 3. 變更 Gamma sterilization 程序,可能導致 PC、PE、PP 或其他塑膠材料產生脆化或變色,使得生物相容性萃取時釋放出更多有毒物質。 |
| 若上述四項評估答案是「Yes」,則很可能須申請新 510(k)。 | 若評估變更有顯著影響,則可能須申請新 510(k)。 |
[Q4. 是否變更包裝 (Packaging) 或失效日期 (Expiration date)?是否用新方法去驗證變更 (Support the change)?]
一般來說,變更包裝或失效日期是不影響原有的 510(k),只要廠商遵循 21 CFR Part 820 進行變更管制即可。但是,若廠商用新的方式 (非原 510(k) 中描述的方式) 去驗證新包裝或新失效日期,則可能須申請新 510(k)。若只是用新版的國家/國際標準進行驗證,仍屬於「使用原 510(k) 中描述的方法」,因此不影響 510(k)。
| FDA 在指引的 P.49 舉例如下: Change: The manufacturer changes the packaging for their device, which is provided sterile, from one variant of polyethylene to another due to a material supplier change. An analysis shows the new polyethylene has no impurities that could affect the device’s biocompatibility. The manufacturer will use the same package integrity test protocol as the one described in its previously cleared 510(k) to support the change. Relevant questions: Q4 – Is there a change in packaging or expiration dating? Yes. Q4 – Is the same method or protocol, as described in a previously cleared 510(k) used to support the change? Yes. Decision: Documentation. — 為了加深 Q4 的印象,所以收錄此範例。 |
[Q5. 有尺寸 (Dimensions)、性能 (Performance)、無線通訊 (Wireless communication)、材料 (Material)、零組件 / 附件 (Components / accessories)、病患或使用者介面 (Patient / user interface) 的變更嗎?]
可能因為客訴、或廠內不合格事項等等原因,廠商想要變更產品。
這部分 (Q5) 包含了所有類型的變更 (不僅限於下列變更),特別說明以下六類變更:
| 變更類型 | 評估重點 (除了這部分的重點外,還須考量本段後面四個重點) |
|---|---|
| 尺寸變更 (Dimension changes) | 要評估所變更的尺寸是否為關鍵尺寸,是否會顯著影響產品安全與功效。 例如,增加內視鏡直徑 1 mm,對於安全與功效的影響可能遠大於增加內視鏡長度 1 mm。 但是,若變更的尺寸已包含在原本 510(k) 的規格內,則此類變更通常不用申請新的 510(k)。 例如,原有 510(k) 許可的內視鏡直徑有兩個型號:2 mm & 4 mm,現在要新增一個型號直徑為 3 mm,則此類變更通常不會顯著影響安全與功效,因此不會到達申請新 510(k) 的閾值。 |
| 性能變更 (Device performance changes) | 本文前面「指導原則一」所描述的,「意圖重大變更者,通常要重申請 510(k),但沒此意圖者,仍須評估是否超過閾值」。 |
| 無線通訊變更 (Wireless communication changes) | 將通訊方式由有線 (Wierd) 變更成無線 (Wireless),因為牽涉到資料傳輸、網路安全等問題,通常會顯著影響安全與功效,尤其當無線通訊的目的是用來控制產品時,更是如此。 上述所指的「通訊」,包含產品零組件之間、或與其他產品間的通訊。 |
| 材料變更 (Material changes) | 此類變更除了影響生物相容性、清潔滅菌等方面外,也要考慮是否會影響 Performance characteristic,例如,是否更容易斷裂、容易滑動? (可參考本指引 第 14.c 範例 (P. 50)) |
| 零組件 / 附件變更 (Components or accessories changes) | 尤其要考慮: 1. 是否會影響到其他零組件 / 附件的使用; 2. 是否會使得產品有新的使用方式; 3. 是否破壞與其他產品或零組件 / 附件之間的相容性; 4. 是否顯著影響產品的風險概況 (Risk profile)。 |
| 病患或使用者介面變更 (Changes in the human factors of the patient / user interface) | 介於產品與病患 / 使用者之間的介面 (例如,顯示 (Displays)、控制 (Controls)、包裝 (Packaging)) 變更時,都須考量對產品的影響。 例如,新增/變更警報、變更控制面板的版面 (可能影響產品在不同使用情境下使用方式)、變更提供資訊給病患 / 使用者的方式 (可能影響解讀)、變更持續性正壓呼吸面罩 (CPAP mask) 與病患的接觸方式、或修改使用流程 (User workflow) 等等,均須考慮是否會影響安全與功效。 通常,為了改善舒適度而針對使用介面的修改是不用申請新 510(k) 的,但是有例外。例如,將手術用具的馬達改得更靠近手持部位或手術部位,可能容易導致手術者或病患燒傷。(可參考本指引 第 19 範例 (P. 53)) (可參考〈Applying Human Factors and Usability Engineering to Medical Devices〉了解更多 FDA 對人因工程的想法) |
| FDA 在指引的 P.50 舉例如下: Change: A biliary stent manufacturer adds a new stent diameter to a family of stents, within the range of the diameters of the manufacturer’s previously cleared stents. The stent lengths are unchanged. The previously cleared 510(k) for the stents objectively demonstrated that the smallest and largest stent diameters (the minimum and maximum ends of the diameter size range) were the worst-case scenarios in terms of the safety and effectiveness risks. Relevant questions: Q5 – Is it any other change in design (e.g., dimensions, performance specifications, wireless communication, components or accessories, or the patient/user interface)? Yes. Q5 – Does the change significantly affect the use of the device? No. Because the new diameter is within the range of the previously cleared stents, the manufacturer determines that the change does not significantly affect the use of the device. Q5 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? No. Since the new stent diameter is within the range of the manufacturer’s previously cleared stents of the same lengths, and the previously cleared 510(k) objectively demonstrated that the smallest and largest diameter sizes represented worst-case scenarios in terms of the safety and effectiveness risks for this stent length, the new diameter would not significantly affect the risk profile of the device. Decision: Documentation. — 節錄此範例是為了說明,「並非」變更的尺寸已包含在原本 510(k) 的規格內,則此類變更「一定」不用申請新的 510(k)。 以上面 Risk-based assessment 為例,因為廠商在前次 510(k) 中已證明最大 / 最小尺寸是 Worst-case,所以,新增的尺寸在最大 / 最小尺寸中間,風險很低。(請參考前面關於「尺寸變更 (Dimension changes)」的筆記) 另可參考本指引的範例 16.(P. 51)。 |
| FDA 在指引的 P.53 舉例如下: Change: The manufacturer adds a foot switch to control an endoscopic electrosurgical unit. The previously cleared device did not have a foot switch. Relevant questions: Q5 – Is it any other change in design (e.g., dimensions, performance specifications, wireless communication, components or accessories, or the patient/user interface)? Yes. This is a change to the device’s user interface. Q5 – Does the change significantly affect the use of the device? No. The addition of a foot switch would not significantly affect the use of the device. Q5 – Does a risk-based assessment of the changed device identify any new risks or significantly modified existing risks? Yes. The risk analysis identified human factors and compatibility risks for the footswitch that did not exist for the previously cleared device. At least some of these risks were associated with the potential for unintentional activation of energy, which could result in a serious harm. Decision: New 510(k). — 此範例中,FDA 評估加入腳踏開關不會顯著影響產品的使用,但會導致一些新的嚴重風險,例如,不小心透過腳踏開關打開電燒。 |
針對上述變更,FDA 列出以下四個評估重點:
重點一、變更是否顯著影響產品使用方法?
廠商須評估此變更是否可能擴展產品的使用族群、讓不同的使用族群使用本產品、讓產品有新的使用目的,進而影響產品的風險概況 (Risk profile)。
例如,這個改變是否導致使用族群從醫生 -> 業餘使用者、家醫科醫師 (General practitioner) -> 外科醫生 (Surgeon)。若是,則可能因影響產品的風險概況而須申請新的 510(k)。
除了考量使用族群,也要評估是否會變更使用環境。例如,醫療院所使用 – > 家用、或救護車上使用,都可能引進新的風險,或顯著影響現有風險。若是這樣,則可能須申請新 510(k)。
關於使用環境,FDA 列出以下評估重點:
| 環境變更 | 評估重點 |
|---|---|
| 溫度、濕度、水、塵 (Soils)、光 | 否會影響儀器的使用 |
| 噪音 | 否會讓使用者忽略警報聲 |
| 周遭其他設備 | 否會有 EMC 問題 (被電磁干擾、或電磁干擾別人) |
| 核磁共振 (MRI) | 是否此項變更將使本產品可能與 MRI 共用 |
重點二、變更是否導致新的風險,或顯著影響現有風險 (Significantly modified existing risks)?
可參考「指導原則四 (納入風險管理概念)」。
重點三、是否須臨床資料 (Clinical data) 來評估產品的安全與功效以完成設計確效 (Design validation?)?
若發現實驗室測試 (Bench test) 或模擬測試 (Simulations) 不足以評估 (Assess) 本次的變更對安全與功效的影響,唯有透過臨床資料才可進行設計確效。此時,很可能須申請新的 510(k)。
上述所指的臨床資料的目的僅限於用來設計確效,而非用來評估使用者 / 病患的喜好。若廠商需臨床資料來評估使用者 / 病患的喜好,則應該不需申請新 510(k)。
重點四、設計驗證 (Verification) / 確效 (Validation) 是否發現任何非預期的安全與功效問題?
請參考「指導原則五 (若 Verification & Validation 出現非預期結果,則可能須申請新的 510(k))」。
若 V&V activities 出現以下情形,代表顯著影響安全與功效,因此很可能這項變更須申請新 510(k):
- 出現非預期的結果、
- 發現安全與功效相關問題、
- 無法符合原本的允收標準,或者
- 原本 (Routine) 的 V&V 不足以驗證 / 確效此次設計變更,需要新方法、新允收標準。
請注意,只是因為有新版的國家/國際標準、允收標準,而使用新方式進行 V&V,則仍屬於「Rutine V&V」,並不影響 510(k);但若不得不用新版的國家/國際標準、允收標準進行 V&V,則很可能這項變更須申請新 510(k)。
FDA 在此指引列出兩個範例:
範例一:變更導尿管 (Urinary drainage (Foley) catheter) 長度
為了更方便連接導尿管到尿液儲存裝置,廠商增長導尿管長度幾 mm。新導尿管長度超過原 510(k) 核准的長度,但廠商評估只是加長一點點並沒有新增任何風險、也沒顯著影響現有風險、安全與功效,暫時判定不影響現有 510(k)。接著廠商驗證新導尿管可以安全有效地運作,沒有出現任何非預期結果。
上述情況,廠商可將設計變更相關評估文件留存備查即可,不需申請新 510(k)。
範例二:監控設備改用更敏感的比較器電路 (Comparator circuit)
廠商想更換監控設備內的比較器電路,以提升敏感度,也因為這項變更,還須變更其它規格。廠商透過計算,初步判定此變更沒有引起新風險或顯著影響現有風險,但後續的 Routine V&V 卻發現了非預期的結果。
此時,廠商須重新仔細、小心地評估,是否有未發現安全或功效的疑慮。
次表 C – Materials Changes (五個評估重點)

除了接下來的評估重點,廠商在變更材料後,還須考慮是否有影響到其他規格。例如,可能因為變更材料,所以須修改 Labeling (刪除 / 新增新的警語、禁忌症),或者降低結構強度等規格。
[Q1. 是否為 IVD (In vitro diagnostic) 產品?]若是,請跳過此部分,直接參考本指引「Section D – Technology, Engineering, Performance, and Materials Changes for In Vitro Diagnostic Devices」。
[Q2. 是否變更材料類型 (Type)、材料配方 (Formulation)、化學成分 (Composition) 或材料加工程序 (Material’s processing)?]
什麼是「材料類型」的變更?
例如,由天然乳膠橡膠 (Natural latex rubber) -> 合成乳膠 (Synthetic rubber) 即屬此類。
什麼是「材料配方」變更?
加入添加物 (Additives)、顏色等企圖改變材料特性或穩定性。例如,由 300 系不銹鋼 (Series 300 stainless steel) 變更為 400 系不銹鋼 (Series 400 stainless steel)。
但像是加工助劑 (Processing aids)、脫膜劑 (Mold release agents)、殘餘汙染物 (Residual contaminants) 等等,這些預期不是材料一部分的東西的變更,不屬於「材料配方」變更 (但應該算變更「材料加工程序」)。若以下類型有變更,則廠商須進行 Q3 的評估:
- 材料類型 (Material type)、
- 材料配方 (Material formulation)、
- 化學成分 (Chemical composition) 或
- 材料加工程序 (Material’s processing)
- 供應商換料、或換供應商 (Changes made by a material supplier, or changes from one supplier to another)
若非上述類型的變更,則應該不需要變更 510(k)。
| FDA 在指引的 P.59 舉例如下: Change: The manufacturer of a dental implant changes the surface of a titanium dental implant from an untreated surface to one that is acid-etched. The surface is in direct contact with the patient’s bone. The manufacturer has not previously used the acid-etching process, and a cleaning process is necessary to remove acid from the device surface. Relevant questions: Q2 – Is this a change in material type, material formulation, chemical composition, or the material’s processing? Yes. The material processing of the device has been changed. Q3 – Will the changed material directly or indirectly contact body tissues or fluids? Yes. Q4 – Does a risk assessment identify any new or increased biocompatibility risks? Yes. The risk analysis identified that the residue from the acid-etching process is a new chemical on the device and introduces a new biocompatibility risk, which may affect the biocompatibility of the device. Q4.1 – Has the manufacturer used the same material in a similar legally marketed device? No. The manufacturer has not previously used the acid-etching process. Decision: New 510(k). — 這算是變更「材料加工程序 (Material’s processing)」的典型範例,但不是只要變更材料加工程序都要申請新 510(k) 喔。 |
| FDA 在指引的 P.59 舉例如下: Change: The manufacturer of an implantable device applies a temporary tape to the device for identification of manufacturing steps. The tape has been demonstrated in peer-reviewed literature to not leave adhesive on the surface of the device. Relevant questions: Q2 – Is this a change in material type, material formulation, chemical composition, or the material’s processing? Yes. The material processing of the device has been changed. Q3 – Will the changed material directly or indirectly contact body tissues or fluids? Yes. Q4 – Does a risk assessment identify any new or increased biocompatibility risks? No. A risk assessment was performed and identified that the tape has been previously demonstrated to not leave adhesive on the surface of the device. Q5 – Could the change affect the device’s performance specifications? No. The tape is temporary for manufacturing purposes, and is removed before clinical use of the device. Since the tape has been demonstrated to not leave adhesive on the surface of the device, it would not be expected to affect the device’s performance. Decision: Documentation. — 雖然有改製程,且被改製程的材料會和身體接觸,不過膠帶只是暫時黏在上面,且已經證實不會有殘留,所以不會有風險,也不會影響 Performance specifications。 |
[Q3. 變更材料會直接或間接接觸身體組織或體液?]
「直接接觸」顧名思義就是「材料會物理性地與病人或使用者接觸」。
「間接接觸」則是「材料先與液體、氣體接觸後,這些液體、氣體再與病人或使用者接觸」(材料本身不會與病人、使用者有物理性地接觸)。
若 Q3 的答案是「Yes」,則繼續評估 Q4;若此答案為「No」,則進入 Q5。
[Q4. 此變更經 Risk-based assessment 發現新的生物相容性疑慮,或增加舊有相關的疑慮?]
FDA 要求廠商以 Risk-based assessment 評估有無:
- 新的生物相容性疑慮,或
- 增加舊有相關的疑慮。
例如,因為這項變更,導致要執行原本不需進行的植入試驗 (Implantation test),此即「新的生物相容性疑慮」;若原本產品就須分析基因毒性 (Genotoxicity analysis),此時新增一個同樣也須測試基因毒性的新材料到產品內,這就是「增加舊有相關的疑慮」。
(參考〈Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process〉以了解如何進行生物相容性的 Risk-based assessment)
若 Q4 評估結果為「無」,那接著進行 Q5 評估;若「有」,則接著評估「Q4.1 是否有合法上市的類似產品使用類似材料?」
不過,若是有經驗的人 (Knowledgeable individual) 評估新舊材料的化學組成 (Chemical composition) 或物理性質 (Physical properties) 差異不大,沒有新的生物相容性疑慮,也可評估結果為「無」,接著進行 Q5 評估。
[Q4.1 是否有合法上市的類似產品使用類似材料?]
若 Q4 評估結果為「Yes」,則要進行 Q4.1 的評估。
此部分評估有以下兩點需注意:
- 都是拿最終成品 (Final finished device) 來比較,而非原料。
- 要和「自己廠」已合法上市 (Own legally marketed devices) 的產品比較 (除非有辦法得知其他廠商產品的確切配方、製程等資訊。)
評估重點如下 (A:本廠已合法上市產品;B:此次變更產品):
- A 、B 有相同配方 (Formulation)、化學成分 (Composition)、製程 (包含滅菌 (Sterilization));
- A 無上市後生物相容性不良事件;
- A、B 接觸方式 (Type of contact) 相同,或 A 相較於 B 有更多高風險的接觸方式 (例如,Mucosal membrane contact > Intact skin contact、Blood contact > Tissue/bone contact);
- A、B 的尺寸、結構形狀 (Geometry)、產品表面特性、預期用途、與其它零組件 / 產品相接方式 (例如,黏著劑類型、熱焊條件) 的差異不影響生物相容性;
- A、B 有相同接觸時間 (Duration of contact),或 A 比 B 有更長的接觸時間。
假如經過上述五點評估,結果均為「Yes」,則進行 Q5 評估;反之,則須申請新 510(k)。
| FDA 在指引的 P.55 舉例如下: Change: The manufacturer of a catheter changes the material of its catheter from polymer A to polymer B. The manufacturer has not previously used polymer B in any of its devices, but knows of another catheter on the market from a different manufacturer with the same cleared indications for use that uses polymer B. Relevant questions: Q2 – Is this a change in material type, material formulation, chemical composition, or the material’s processing? Yes. Q3 – Will the changed material directly or indirectly contact body tissues or fluids? Yes. Q4 – Does a risk assessment identify any new or increased biocompatibility risks? Yes. Polymer B has a different chemical formulation than polymer A. The risk assessment identifies that the new formulation presents a new biocompatibility risk. Q4.1 – Has the manufacturer used the same material in a similar legally marketed device? No, the manufacturer has not used the same material before. Even though there is another catheter from a different manufacturer on the market made of polymer B, the other device may have a different formulation or different manufacturing or finishing processes that could affect the biocompatibility or performance. Decision: New 510(k). — 此範例說明了,就算知道其他廠也有用一樣的材料,也不可作為證明安全與功效的依據。畢竟,影響材料的因素很多,例如,添加物、製程,而一般是無法確定這些因素,他廠是否與本廠一樣。 |
| FDA 在指引的 P.56 舉例如下: Change: A manufacturer changes the material of its catheter, intended for prolonged blood contact, from polymer A to polymer B. The manufacturer has used the same polymer B in another cleared device; however, this other device was indicated for a use with limited duration and skin contact only. Relevant questions: Q2 – Is this a change in material type, material formulation, chemical composition, or the material’s processing? Yes. Q3 – Will the changed material directly or indirectly contact body tissues or fluids? Yes. Q4 – Does a risk assessment identify any new or increased biocompatibility risks? Yes. Polymer B has a different chemical formulation than polymer A. The risk assessment identifies that the new formulation presents a new biocompatibility risk. Q4.1 – Has the manufacturer used the same material in a similar legally marketed device? No. The manufacturer has used the same polymer B, with the same formulation and processing, in another device, however, the other device was subject to a less risky type and duration of contact. The modified device will be subjected to additional biocompatibility risks compared to the other polymer B device, and therefore the use of polymer B in the other device does not address the biocompatibility concerns identified in the risk assessment covered in C4. Decision: New 510(k). — 雖然在此範例中,廠商已有使用 B 材料的上市產品,但 B 材料產品與病患接觸方式為 Skin contact only (原 A 材料產品接觸方式為 Blood path, indirect),相較於原 A 材料產品的接觸時間又比較短。因此,如前面「評估重點」,不可判定 Same material in a similar legally marketed device。 |
[Q5. 變更是否會影響產品的性能規格 (Performance specification)?]
評估變更材料是否會影響到產品的機械性質 (Mechanical properties),例如:強度 (Strength)、硬度 (Hardness),進而影響性能規格。也須考量新材料是否會被原本的滅菌 (Sterilization)、清潔 (Cleaning)、消毒 (Disinfection) 影響。
若評估是「Yes」,則須回 Q5 進一步評估尺寸、性能、使用者介面等等因素對安全與功效的影響。
若評估結果是「No」,則此變更應該沒超過影響現有 510(k) 的閾值。
| FDA 在指引的 P.58 舉例如下: Change: A manufacturer decides to change the material of a catheter from material A to material B. Material B is used in another of the manufacturer’s own cleared catheters, which has the same type and duration of patient contact, but different performance specifications. Both materials A and B are molded and are sterilized by ethylene oxide. Relevant questions: Q2 – Is this a change in material type, material formulation, chemical composition, or the material’s processing? Yes. Q3 – Will the changed material directly or indirectly contact body tissues or fluids? Yes. Q4 – Does a risk assessment identify any new or increased biocompatibility risks? Yes. Material B has a different chemical formulation. The risk assessment identifies that the new formulation presents a new biocompatibility risk. Q4.1 – Has the manufacturer used the same material in a similar legally marketed device? Yes. The manufacturer has used material B in another cleared catheter, and the processing is the same. In addition, the size and geometry of the new device would not affect curing of the polymer or result in more material in the new device, and there are no differences in how material B is joined to other components of the catheter (e.g., type of adhesive, or conditions of heat welding) that could result in different interactive chemistries. Q5 – Could the change affect the device’s performance specifications? Yes. The manufacturer used the same material B in another model of catheter; however, the performance specifications were different. The new material could potentially affect the device’s performance, so the manufacturer is directed to 次表 B Q5. 次表 B Q5 – Is it any other change in design (e.g., dimensions, performance specifications, wireless communication, components or accessories, or the patient/user interface)? Yes. 次表 B Q5 – Does the change significantly affect the use of the device? No. The new material does not significantly affect the use of this device. 次表 B Q5 – Does a risk assessment of the changed device identify any new risks or significantly modified existing risks? If the new material has significantly different physical properties than the material in the previously cleared device, the risk profile of the device could be significantly affected in terms of risk score, risk acceptability, etc., and submission of a new 510(k) may be required. However, for the purposes of this example, the new material is not expected to have significantly different physical properties, so a 510(k) would not be required. Decision: Documentation. — 節錄此範例是因為,原來,變更產品和廠內類似品的 Performance specifications 雖然不同: 1. 仍算是「The manufacturer used the same material in a same material in a similar legally marketed device」。 2. 但在評估「Q5 – Could the change affect the device’s performance specifications?」時,要考慮更換材料是否會影響到產品的機械性質 (Mechanical properties),例如:強度 (Strength)、硬度 (Hardness),進而影響性能規格。 |
次表 D – Technology, Engineering, Performance, and Materials Changes for In Vitro Diagnostic Devices
此章節專用於 IVD 產品,故先跳過待之後有時間再補充。
關於軟/韌體的變更
背景介紹
有別於〈General modifications guidance〉用於評估一般變更,〈Software-specific modifications guidance〉專門用於評估軟 / 韌體的變更。兩者雖然著重的面向不同,但都是為了符合 21 CFR 807.81(a)(3) 針對「上市後產品變更達一定程度後須申請新 510(k)」的要求,協助廠商與 FDA 雙方在解讀「Could significantly affect the safety or effectiveness of the device」可以趨向一致,進一步協助廠商:
1. Analyzing how changes in devices may affect safety or effectiveness, and
Software-specific modifications guidance | II. Background
2. Determining whether a new 510(k) must be submitted for a particular type of change.
一句話來總攝〈Software-specific modifications guidance〉的目的如下:
This guidance will assist industry and Agency staff in determining when a software (including firmware) change to a medical device may require a manufacturer to submit and obtain FDA clearance of a new premarket notification (510(k)).
oftware-specific modifications guidance | I. Introduction
| 當變更除了影響軟體外,也影響到 Labeling 或硬體的話,廠商須同時參考〈General modifications guidance〉和〈Software-specific modifications guidance〉進行評估。若評估任一變更程度到達「申請變更閾值」,就須申請新的 510(k)。 |
| 品質系統 (21 CFR Part 820、ISO 13485 等) 對於變更管制的要求,以及 21 CFR 807.81(a)(3) 的背景介紹,已在前面摘要重點過了,故〈關於軟/韌體的變更〉章節將跳過〈Software-specific modifications guidance〉對這部分的解釋。 |
關於本指引所謂的「Software」,FDA 定義如下:
1) Set of electronic instructions used to control the actions or output of a medical device,
Software-specific modifications guidance | III. Scope
2) to provide input to or output from a medical device, or
3) to provide the actions of a medical device.
所以,包含:
1) software that is embedded within or a component of a medical device,
2) software that is an accessory to another medical device, or
3) software that is intended to be used for one or more medical purposes that performs these purposes without being part of a hardware medical device. (也就是 SaMD 啦)
Software-specific modifications guidance | III. Scope
| 本指引不適用、沒涵蓋: 1. 不適用非軟體的變更 (非軟體變更請參考「關於軟/韌體以外的變更」)。 2. 不適用於那些不須遵守法規的軟體。例如,被 FDA 判定不屬於醫材的 Mobile App (參考〈Policy for Device Software Functions and Mobile Medical Applications〉)。 3. 沒涵蓋軟體生命週期管理,相關管理規範請參考〈AAMI/ANSI/IEC 62304〉,所須產出的 510(k) 軟體文件請參考〈Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices〉。 4. 沒涵蓋軟體確效 (相關指引請參考〈General Principles of Software Validation〉)。 |
十大指導原則
本指引的指導原則基本上和〈General modifications guidance〉的「十大指導原則」完全一樣,以下僅列出兩本指引的差異 (但差異也不大,僅是特別為軟體特性些微修改):
| 要記得搭配〈General modifications guidance〉的「十大指導原則」一起看喔! |
指導原則三、注意是否有非預期的影響
本指引舉例,更新作業系統 (Operating system,OS) 時可能會非預期地影響產品的驅動程式、或其他程式的相容性。廠商要完整評估預期、非預期的變更所造成的影響是否須申請新 510(k)。
指導原則四、納入風險管理概念
本指引的專有名詞除了引用自〈ISO 14971〉外,也引用〈IEC/TR 80002-1: Guidance on the application of ISO 14971 to medical device software〉。IEC/TR 80002-1 我沒看過,依 FDA 這部分的解釋,似乎因為軟體的失效機率難以用傳統 ISO 14971 的方式估計,因此須著重在危害 (Harm) 本身 (If the overall probability of occurrence of harm cannot be estimated, the estimation of risk should be based on the severity of harm alone)。
指導原則六、須考量同時的個別變更,也須將這些變更綜合評估
本指引的特別說明,「for software, each individual line change in the code may not constitute an individual change in the device.」
指導原則九、若因變更須申請新 510(k),則送審文件中須描述其它次要變更
本指引的特別說明,「Manufacturers should know which versions of off-the-shelf software and/or firmware are included in their device even if that level of detail is not included in a 510(k).」
正式進入評估流程
與〈General modifications guidance〉一樣,本指引目的是協助廠商評估變更對於安全與功效的影響。同樣地,變更程度大小要和「Original device」相比,若超過 21 CFR 807.81(a)(3) 定義的閾值 (無論正 / 負面影響),
- Could significantly affect the safety or effectiveness of the device, 或有
- Major change or modification in the intended use of the device,
就須申請新 510(k)。

[Q1. 此變更只為了加強網路安全 (Cybersecurity),對產品或軟體不會有其他的影響?]
若只為了加強網路安全所執行的變更,通常只要透過必要的分析、驗證 (Verification)、確效 (Validation) 確認沒有影響產品安全 (Safety) 與功效 (Effectiveness),此類變更不需申請新 510(k)。
但若這類的變更對於網路安全以外的部分也有非預期的影響,則須繼續接下來的評估 (Q2、Q3、Q4)。關於網路安全,可參考〈Content of Premarket Submissions for Management of Cybersecurity in Medical Devices〉和〈Postmarket Management of Cybersecurity in Medical Devices〉。
[Q2. 變更僅為回復系統到最近一次許可產品 (Most recently cleared device) 的規格?]
若僅為了回復系統到最近一次許可產品的規格 (可能先前的設計文件上有規劃某軟體規格,但忘記加入),則這類變更不需申請新 510(k),但廠商仍須確認這類變更沒有影響到性能規格 (Performance specification)。
| FDA 在本指引的 P.20 舉例如下: Change: An issue was observed for an IVD analyzer in the field. The IVD analyzer software collects reagent administrative records (e.g., material number, lot number, expiration date). The records are to be written by the software into a database table. After enough records are collected to fill the table, newly-collected records are then to be written in the first row of the table, overwriting previous records. Under certain conditions, the software system mistakenly merges the new data with the existing data in the first row of the table in the database, which may lead to an incorrect result. The cause of the bug was found to be an incorrectly worded software requirement that led to an error in the software code. The requirement was rewritten. An additional software change was made to correct the software code in the control unit of the analyzer. Code was modified to ensure that data written to a database is not merged with any existing data. The change to the software involved creating an entirely separate database within the instrument software, specifically for the administrative records stored for reagents to prevent records from being merged. This change required a specification change at the unit level to describe the new database. Relevant questions: Q2 – Is the change made solely to return the system into specification of the most recently cleared device? No. A change was made to correct a coding error by adding a new database. This caused a change to the design specifications of the software. Outcome: Continue to other decision points. — 這個變更「看似」是回復系統到最近一次許可產品的規格,實際卻修改了 Software requirement specification (SRS),不算 Q2 所描述的情況,故須繼續評估其他要點。 |
| FDA 在本指引的 P.19 舉例如下: Change: An issue was observed in IVD analyzer software that collects reagent administrative records (e.g., material number, lot number, expiration date). The records are to be written by the software into a database table. After enough records are collected to fill the table, newly-collected records are then to be written in the first row of the table, overwriting previous records. Because of a software bug, the system mistakenly merges the new data with the existing data in the first row of the table. The cause of the anomaly was determined to be a coding error that did not affect any of the software requirements. A change was made to correct the software code in the control unit of the analyzer to ensure that data written to a row in the table is not merged with any existing data. The change to the software involved modification of a table within the analyzer software to add new columns to track the administrative data stored for reagents to prevent data from being merged. Relevant questions: Q2 – Is the change made solely to return the system into specification of the most recently cleared device? Yes. The change was only to address a software anomaly and was not a change in specification or functionality of the most recently cleared device. Outcome: Document the change to file. — 和上個範例一樣,都是輸入過多資料後,後輸入的可能會覆蓋已輸入的資料。但為何後面這項變更只要將變更紀錄留存,但前者卻須繼續評估其他要點呢? 差別在於,前者原本的規格 (SRS) 就寫錯了,導致資料被覆寫,為了矯正這個問題,須修改規格,因此不屬於「回復系統到最近一次許可產品的規格」(而是修改規格);後者,原本的規格沒寫錯,只是原本程式碼寫錯了,僅修改程式碼後就可矯正此問題,屬於「回復系統到最近一次許可產品的規格」。 |
[Q3. 變更是否導致新的風險 (Risk),或修改現有風險而可能導致嚴重危害 (Harm),且此類風險並未於最近一次許可產品中被有效地降低?]
| 我將本指引的第三個評估重點,「What are the impacts of any changes to risks associated with use of the device and the impacts of any changes to the risk controls for the device?」下面兩個子問題,獨立成本文的 Q3、Q4。 |
「嚴重危害」、「成因」在此定義為:
Significant harm refers to situations where the risk level is serious or more severe, e.g., the risk could result in injury or impairment requiring professional medical intervention, permanent impairment, or death.
The term “cause” refers to one possible component in the “sequence of events,” that can lead to a hazardous situation and possible harm, as described in ISO 14971.
Software-specific modifications guidance | V. How to Use This Guidance, 3.a)
若變更符合以下所有條件,則須申請新 510(k):
- 導致新的、或修改現有的危害 (Hazard)、危害情況 (Hazardous situation)、或成因 (Cause)。
- 上述的情況 (第 1 點) 導致的危害 (Harm) 等級,在控制風險前 (Pre-mitigation) 達到「Serious」或「More severe」程度。(請參考「Significant harm」的定義)。
- 危害 (Hazard)、危害情況 (Hazardous situation)、或成因 (Cause) 未在最近一次許可產品 (Most recently cleared device) 中被有效地降低至可接受的程度。
| FDA 在本指引的 P.21 舉例如下: Change: The manufacturer makes a software modification to prevent a patient sample probe motor from overheating during a customer maintenance procedure. Power is applied to the sample probe motor to keep the sample probe assembly in a locked position during the user maintenance procedure. In the field, it was reported that applying power to the sample probe motor for more than 20 minutes causes the motor to overheat and creates a potential minor burn hazard (i.e., it becomes too hot to touch safely). The software change applies a timeout to power being applied to the sample probe motor during the maintenance procedure; after ten minutes, power to the sample probe motor is turned off. An additional software change adds a message window at the beginning of the procedure to alert the user that the procedure must be completed within a ten-minute window or the system will cut power to the motor. A limit of ten minutes was determined to keep the motor from overheating to the point of creating a potential minor burn hazard. Relevant questions: Q3 – Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? No. The change provides a mitigation to an existing hazardous situation that was not appropriately mitigated in the cleared device. However, the hazardous situation could not cause significant harm. Outcome: Continue to other decision points. — 雖然這項變更屬於修改「現有」的危害情境,也沒有在最近一次許可產品中被有效地降低至可接受的程度,但是此危害情境導致的危害程度,在控制風險前並未達到「Serious」或「More severe」程度,所以只需繼續評估接下來的要點。 |
| FDA 在本指引的 P.22 舉例如下: Change: The device is an implantable, automatically activated monitoring system that records subcutaneous electrocardiograms designed to record the arrhythmias in a patient. The manufacturer has made a software modification to add an alternative programming mode to change the way the device interacts with the programmer. This new programming mode provided different capabilities for data programming, interrogating, and managing the device data and function. The mode introduces new technology that impacts the safety profile of the device as a result of the energy transfer that occurs during programming. Relevant questions: Q3 – Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? Yes. This feature introduces new risks based on the new programming mode that could cause significant harm as a result of energy transfer to the patient. Outcome: Submit the change in a new 510(k). — 雖然這項變更和上個範例不同點在於,這項變更除了造成新的風險外,且此風險在風險控制前的危害等級達到 Significant harm,因此須申請新 510(k)。 (PS.) Significant harm refers to situations where the risk level is serious or more severe, e.g., the risk could result in injury or impairment requiring professional medical intervention, permanent impairment, or death. |
[Q4. 此變更是否針對可能的嚴重危害 (Significant harm),導入新的風險控制措施 (Risk control measure) 或修改現有的風險控制措施?]
此類變更可能目的就是修改原有的風險控制措施,又或者因為此類變更而須導入新的 / 修改現有的風險控制措施,無論哪種,只要新增 / 修改與嚴重危害相關的風險控制措施,都須申請新 510(k)。
但若新增 / 修改的風險控制措施,僅是下列情況,則不需申請新 510(k):
- 為了加強 (Enhancing) 最近一次許可產品中已有的風險控制措施;或
- 新增最近一次許可產品中已有的風險控制措施的備援措施 (Redundant risk control measures in the most recently cleared device)。
| 以下是 IEC/TR 80002-1:2009 對「Redundancy」的定義 (Section 2.2): “provision of multiple components or mechanisms to achieve the same function such that failure of one or more of the components or mechanisms does not prevent the performance of the function.“ |
| FDA 在本指引的 P.23 舉例如下: Change: The device is a robotically assisted surgical system that utilizes position sensors. The system incorporates primary and secondary sensors to monitor the movement of actuators to prevent uncontrolled motion of the instrument in the event of a component failure. The system goes into a fault state and halts motion if the position information between the sensors does not match within a certain threshold. The threshold for each actuator is programmed in the software and there is a specification for how much overall movement is acceptable at the tip of the instrument before movement stops. The manufacturer makes a software change to the threshold settings for the position sensors; specifically, the software specification that defines the tip movement was widened and the software was changed to allow the wider tolerance. The change was made to minimize false assertion of the safety system, and the change in the specification for movement at the tip of the instrument was still within an appropriate safety tolerance for the device, as determined by analysis done by the manufacturer. However, the change modified an existing risk control (distance that can be traveled under fault conditions) that could significantly affect safety or effectiveness. Relevant questions: Q4 – Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? Yes. The modified threshold values do not meet the specification for overall tip movement, which was required in the most recently cleared device to effectively mitigate the hazardous situation that could result in significant harm. Thus, the change necessitated modification of an existing risk control in the most recently cleared device and submission of a new 510(k) is required. Outcome: Submit the change in a new 510(k). — 這變更屬於 1) 修改現有與嚴重危害相關的風險控制措施,且 2) 前次 510(k) 有審查這項風險控制措施。因此這次變更須申請新 510(k)。 |
| FDA 在本指引的 P.23 舉例如下: Change: The device is a robotically assisted surgical system that utilizes position sensors. The system incorporates primary and secondary sensors to monitor the movement of actuators to prevent uncontrolled motion of the instrument in the event of a component failure. The system goes into a fault state and halts motion if the position information between the sensors does not match within a certain threshold. The threshold for each actuator is programmed in the software and there is a specification for how much overall movement is acceptable at the tip of the instrument before movement stops. The manufacturer makes a software change to the threshold settings for the position sensors; specifically, the software was modified to better calculate overall movement. The change was made to minimize false assertion of the safety system, which required the surgeon to hit an override button to continue. This requirement can be a nuisance and distract from surgery. The modified software continued to meet the specification for movement at the tip of the instrument after a component failure. Relevant questions: Q4 – Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? No. This change modifies sensor threshold parameters so that transient conditions that can be present during normal operation do not cause unnecessary activation of the risk control measure. The change makes the system more noise-tolerant without impacting true positive detection for the risk control measure. The overall movement criteria are met under all fault conditions. Outcome: Continue to other decision points. — 在此範例中,廠商透過改善軟體對機器移動的計算,降低誤啟動風險控制措施的機率。這屬於「加強最近一次許可產品中已有的風險控制措施」的變更,所以只需繼續評估接下來的要點。 |
| FDA 在本指引的 P.24 舉例如下: Change: A PACS provides the option to print images along with a copy of the diagnostic findings from the radiologist. There is data on each page allowing the user to match each page to the corresponding information (e.g., patient ID, Study Identifier). This data helps to address the known risk of pages being mixed-up after printout. Based on customer preference, the manufacturer decided to enhance this existing risk control and have actual patient information and demographics printed on each page. This is intended to be easier for the user to identify which pages belong together and, as a result, further decrease the risk of mixing up printed pages. Relevant questions: Q4 – Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? No. The risk is already sufficiently mitigated with the original risk controls (that is, to have patient identification related information on each printed page). This software modification is a redundant risk control that was not made in response to a new, modified, or previously unknown hazardous situation or cause thereof. Outcome: Continue to other decision points. — 原本的風險控制措施已可有效降低風險,現在保留原有的措施 (每頁報告都有 Patient ID 或 Study Identifier),進一步改善措施 (每頁報告都有完整的病人資訊),這屬於「新增最近一次許可產品中已有的風險控制措施的備援措施」,所以只需繼續評估接下來的要點。 |
| FDA 在本指引的 P.25 舉例如下: Change: A general purpose infusion pump has one alarm to alert the user when an occlusion has been detected. The software change modifies the existing alarm to provide two alarms related to occlusion: occlusion downstream and occlusion upstream. These alarms provide specific information to help resolve the occlusion. Relevant questions: Q4 – Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? Yes. The change modifies the risk control, i.e., the alarm, which is already present for occlusion. This risk control is necessary to improve safety by effectively mitigating specific occlusion events that could result in significant harm if not resolved correctly. Outcome: Submit the change in a new 510(k). — 可能會不小心把此類變更歸類成「加強最近一次許可產品中已有的風險控制措施」的變更,但其實這是屬於「修改與嚴重危害相關的風險控制措施」,因此須申請新 510(k)。 |
[Q5. 變更是否直接顯著影響與預期用途 (Intended use) 相關的臨床功能 (Clinical functionality) 或性能規格 (Performance specifications) 呢?]
| Q5 處理的問題「並非變更 Indications for use、Intended use」,而是在討論「其餘的變更」對 Indications for use、Intended use 的影響。 關於「變更 Indications for use、Intended use」的評估,請參考前文「次表 A – Labeling Changes (九個評估重點)」。 |
因為軟體特性關係,軟體改版頻率相較於硬體較高,但不是所有軟體改版都會影響原本的 510(k) 許可。
FDA 要求廠商特別須評估那些會「顯著影響產品對於預期臨床表現 (Could influence device’s ability to clinically perform as intended)」的軟體變更。
有哪些變更可能會影響預期臨床表現呢?
FDA 舉例如下:Speed、Strength、Response times、Throughput、Limits of operation、Reliability、Delivery rate、Assay performance。
若軟體變更的規格屬於「直接顯著影響與預期用途相關的臨床功能或性能規格 (Significantly affect clinical functionality or performance specifications that are directly associated with the intended use)」,很可能因此變更而須申請新 510(k)。
| 若是 in vitro diagnostic devices (IVDs),則須評估軟體變更對 Clinical decision-making 的影響。(本文暫時跳過指引內關於 IVDs 的部分。) |
| FDA 在本指引的 P.25 舉例如下: Change: A manufacturer makes a software performance enhancement to improve sample throughput time by 20%. Software modifications include changes to decrease assay cycle times by allowing for shorter sample reaction incubation times. Decreasing sample assay times could have an impact on run performance and/or assay performance in a manner that could have a negative impact on diagnosis or therapy delivered to patients. Relevant questions: Q5 – Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? Yes. The change is to increase the throughput performance specification, but has a significant impact on the performance of the device. There is a shorter reaction incubation time and therefore a potential significant impact on diagnostic utility and effectiveness. Outcome: Submit the change in a new 510(k). — 降低樣品反應時間對於產品預期用途相關的產品性能可能有負面影響,因此須申請新 510(k)。 |
| FDA 在本指引的 P.26 舉例如下: Change: A manufacturer makes a software modification to improve sample throughput by 5% by decreasing pre-analytic processing time. Software modifications include a change to decrease sample delivery time from the sample load area to the sample aspiration area. As described here, decreasing sample delivery times do not have an impact on assay performance. Relevant questions: Q5 – Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? No. The modifications do not impact assay performance as it relates to intended use. Improvement resulted from technical analysis of the sample delivery algorithm to optimize timing and remove unnecessary timing delays. Outcome: Document the change to file. — 同樣都是加快分析時間,為什麼前個範例要申請新 510(k),這個範例不需要呢? 因為這個範例僅是透過減少樣品從放置區到吸取區的時間,並不會直接顯著影響與預期用途相關的臨床功能或性能規格,所以只要文件化變更評估過程即可。 |
| FDA 在本指引的 P.27 舉例如下: Change: A sterilizer display provides vital information on the temperature, the pressure, and the remaining cycle time. Software changes are made to increase the font size of these parameters on the display due to customer feedback (not related to any adverse events). The items are all in the same location and the appearance is unchanged aside from the larger font size. Relevant questions: Q5 – Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? No. Since the information was previously displayed, the change has no significant effect on the functionality or the performance of the device. Outcome: Document the change to file. — 只加大字型,沒有新增 / 減少任何資訊,甚至連展現資訊的位置都一樣,所以當然不用申請新 510(k)。 若軟體的使用者介面大幅改版,可能影響 Usability,雖然顯示的資訊一樣,是否也會得到一樣的結論呢? 我不知道,不確定是否要參考〈General modifications guidance〉的 Q5 去評估尺寸、性能、使用者介面等變更所造成的影響。 |
| FDA 在本指引的 P.28 舉例如下: Change: A manufacturer makes a software modification to allow users to silence a low-risk alarm on a dialysis system. The change consists of a “snooze” button that silences the alarm for a set amount of time before resounding. Relevant questions: Q5 – Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? No. The silencing of a non-critical alarm does not impact the clinical functionality. The criteria for the alarm are unchanged from the most recently cleared device. Outcome: Document the change to file. — 因為這個變更是針對「Low-risk alarm」,不會顯著影響臨床功能,故不用申請新 510(k)。 |
常見軟體變更類型
除了「正式進入評估流程」的五個 Q 外,FDA 還列出下列額外考量重點,希望廠商在評估變更影響程度時要納入考量。
有時廠商變更軟體,並非是想要修改什麼功能,而是「Code maintenance」或「Infrastructure modifications」。這時,廠商可能會評估此變更為「沒有影響功能」。但軟體變更對於產品安全與功效的影響常常無法輕易釐清,因此,廠商也須考慮是否有「非預期的影響」。
FDA 希望廠商可以由「Overall architecture of the software」去評估變更的影響。
例如,若軟體架構 (Software architecture) 是有規劃地 (Planned)、模組化 (Modular) 的形式,則變更對其他部分所造成的非預期影響可能性 (Likelihood of unintended impact to other areas of the code may) 就會大幅降低;反之,若軟體架構鬆散 (Looser construct),也沒有清楚的規劃 (Without a clear architectural plan),因為無法明確界定程式碼所影響的範圍,導致非預期影響的可能性就會很大。
FDA 也鼓勵廠商與他們討論「灰色區域」,以釐清「Code maintenance」、「Infrastructure modifications」對安全與功效的影響,進一步準確評估變更是否達到須申請新 510(k) 的閾值。
FDA 整理出六種軟體變更類型,協助廠商可以更完整評估軟體變更對產品的影響。
類型一、Infrastructure 變更
變更「Software support system」、「Software driver」、「OS」均屬於此類型。例如,變更編譯器 (Compiler)、程式語言 (C to C++、C++ to Java)、驅動程式或函式庫 (Library)。
FDA 建議,由「Complexity」的角度來評估此類變更對安全與功效的影響。例如,
- C -> C++,因語法類似 (或其他類似特性,例如 Coding paradigm),可能不用大幅度改寫;
- 函數型 (Functional) 或邏輯型程式語言 (Logical coding paradigm) -> 物件導向程式設計 (Object Oriented Programming paradigm,OOP),加上 C -> C++,這種變更很可能須大幅改寫程式,因此很可能達到須申請新 510(k) 的閾值;
- 變更 Verification / Validation scripts 可能代表大幅變更 Infrastructure,所以 FDA 建議廠商要特別確認這類的變更是否須申請新 510(k)。
類型二、Architecture 變更
變更軟體以支援新的硬體或中介軟體 (Middleware) 均屬此類。這類變更可能會大幅影響產品的效能,或擴張使用環境。
類型三、Core algorithm 變更
這類變更極有可能影響到預期用途 (Intended use),即使變更前後宣稱的性能 (Performance) 是一樣的,或不影響風險概況 (Risk profile),仍可能因為改寫規模很大,因此須申請新 510(k)。
例如,修改監視器的警報演算法、輸液幫浦 (Infusion pump) 馬達控制演算法、或 IVDs 的偵測模組 / 量測模組演算法,均屬此類變更。
類型四、Clarification of Requirements (No Change to Functionality) 變更
此類變更的目的是,(在產品上市後) 使軟體要求 (Software requirements) 更明確。
在沒有變更 / 新增新功能的前提下,修改現有軟體要求的措辭 (Phrasing)、或增加新要求,都屬於此類型。這類型變更大多不需申請新 510(k)。
類型五、Cosmetic Changes (No Change to Functionality) 變更
僅修改外觀 (Appearance)、沒影響臨床使用的變更,均屬此類。
例如,修改軟體每個畫面中的公司 Logo,即使這類修改會影響到很多模組,但此類變更通常不會顯著影響產品的安全與功效,所以不太可能需要申請新 510(k)。
類型六、Reengineering & Refactoring 變更
「Reengineering」、「Refactoring」是兩個常見的軟體維護手法。
一樣要評估變更對規格、性能、風險控制等影響程度,以決定是否要申請新 510(k)。
若,
- 在規格內,為了改善維護性 (Maintainability) 所進行的次要修改,不太可能會影響到現有的 510(k);
- 大幅度改寫程式,很可能須申請新 510(k)。
本指引還說明了「Reengineering」、「Refactoring」的差異:
軟體再工程 (Software reengineering) is defined as the examination and alteration of software to reconstitute it in a new form, and includes the subsequent implementation of the new form. It is often undertaken to replace aging legacy software.
軟體重構工程 (Software refactoring) (也可參考〈什麼是Refactoring?〉) is a disciplined technique for restructuring a software program’s internal structure without changing its clinical performance specification. Refactoring seeks to improve a program structure and its maintainability.
In general, reengineering often results in broader and more complex changes, while refactoring is often narrower in scope and less complex.
Software-specific modifications guidance | Common Software Change Types