筆記目錄
1. 前言 (廢話)
風險管理一直都是很難的部分,無論在人生,或者在醫材領域。雖然 ISO 14971 一直以來都說「殘餘風險高到不可接受,也沒其他風險控制手段時,要評估其利益是否大於風險」:
If a residual risk is not judged acceptable using the criteria established in the risk management plan and further risk control is not practicable, the manufacturer may gather and review data and literature to determine if the benefits of the intended use outweigh this residual risk.
ISO 14971:2019 | 7.4 Benefit-risk analysis
但是,這部分 (其實整個風險管理) 一直是個藝術啊!一般在「做」風險管理文件時,大家都會讓 Residual risk 落在可接受的範圍內,所以超少情況需要進行 Benefit-Risk evaluation。
如果真的要評估呢?好吧~就努力亂掰,努力堆砌華麗文字直到某個程度後驟下結論:「The benefit of using a XXX device outweighs the residual risk of YYY.」
(一直這樣總覺得好膩又好廢啊啊~) 但我就好奇這部分怎樣才能做好呢?
而且,逃得了和尚逃不了廟 (風險管理不用評估,臨床評估也會遇到利弊權衡),Clinical evaluation 中也要探討產品的 Risk/benefit profile、Benefit-risk ratio、Benefit/risk determination。
所以,本篇筆記整理了下列相關指引、網路文章,希望在了解不同時期 (上市前、審查中、上市後)、單位 (顧問公司、官方單位) 對於利弊權衡的想法 / 作法後,(似乎沒時間整理那麼多筆記了 ORZ 只看了 510(k)/PMA/De Novo) 未來我們都可以論述出一個更好 (不心虛) 的 Benefit-Risk evaluation:
- 〈Benefit-Risk Factors to Consider When Determining Substantial Equivalence in Premarket Notifications (510(k)) with Different Technological Characteristics〉(FDA | SEP 2018)
- 〈Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications〉(FDA | AUG 2019)
2. 上市前審查的 Benefit-Risk evaluation 怎麼做?
2.1 關於 510(k) 的 B-R Evaluation

2.1.1 前言 (不是廢話)
這部分是參考於 September 25, 2018 發行的〈Benefit-Risk Factors to Consider When Determining Substantial Equivalence in Premarket Notifications (510(k)) with Different Technological Characteristics〉(後面簡寫為〈510(k) B-R assessment guidance〉) 而成的筆記。
| CDE 在 2014/07/15 有整理前一版指引的重點,大家可參考看看〈美國 FDA 於 2014 年 7 月發表「評估不同技術特點產品之實質等同性所考量的利益與風險因素」指引草案〉。 |
510(k) 上市途徑的中心思想就是「既然這個新醫療器材和某特定已上市醫材 (Predicate) 夠類似 (SE),那安全與功效應該沒問題」。換句話說,透過證明實質相等性 (Substantial equivalence, SE) 可保障新產品有一定程度的安全與功效,而不用勞師動眾靠臨床試驗證明。
雖說申請上市的醫材要和 Predicate 相似,但並不需要一模一樣 (Identical):
The 510(k) premarket review standard (i.e., SE of a new device when compared to a predicate device) does not require a new device to be identical to a predicate device.
510(k) B-R assessment guidance | III. Scope
所以,可能新醫療器材和 Predicate 只有 Intended use 相同,其他特徵,例如,Indication for use、Technological characteristics 不同,仍可被判定為 SE 而取得 510(k) clearance。
同樣地,新醫材的「Benefit-risk profile」也不用與 Predicate 完全相同才可視為 SE。
這篇〈510(k) B-R assessment guidance〉就是在討論「當新醫材的 Benefit-risk profile 與 Predicate 不同時,該如何評估是否為 SE」。
更準確地說,本篇指引是在討論,當在評估 SE 時,若發生以下兩個情況,到底該判定為 SE 或 NSE (Non-substantial equivalence, NSE):
- Increase in risk and increase or equivalent benefit;
- Decrease in benefit and decrease or equivalent risk。
當上面這兩種情況發生時,就要執行 Benefit-risk assessment,以確定「whether the new device is “as safe and effective” as the predicate device」。
| 如想了解實質相等性 (Substantial equivalence, SE) 的評估流程,請參考以下筆記:〈如何使用 Multiple Predicates 來證明 Substantial Equivalence〉與〈如何用 Reference Devices 證明 510(k) 的實質相等性 (SE)〉。 |
2.1.2 SE 的法源依據
首先,要確認新產品在美國是當作醫療器材來管,也就是說,要符合 FDA 的醫療器材定義 (FD&C Act, Sec. 201(h))。否則,可能忙了老半天,結果新產品在美國根本不當作醫材來列管。
接者,確認此醫材的上市途徑是 510(k),而非其他路徑,例如,Listing 或 PMA。既然上市途徑確認是 510(k),必定要挑選一個 Predicate。
有了 Predicate,就要證明新醫材與 Predicate 兩者間的實質相等性 (SE)。SE 定義於 FD&C Act, Sec. 513(i)(1)(A) (21 USC 360c(i)(1)(A)):
For purposes of determinations of substantial equivalence under subsection (f) and section 520(l), the term ‘substantially equivalent’ or ‘substantial equivalence’ means, with respect to a device being compared to a predicate device, that the device has the same intended use as the predicate device and that the Secretary by order has found that the device 一
(i) has the same technological characteristics as the predicate device, or
(ii) (I) has different technological characteristics and the information submitted that the device is substantially equivalent to the predicate device contains information, including appropriate clinical or scientific data if deemed necessary by the Secretary or a person accredited under section 523, that demonstrates that the device is as safe and effective as a legally marketed device, and (II) does not raise different questions of safety and effectiveness than the predicate device.”
整理上段 SE 的規定如下:
- 新醫療器材與 Predicate 要有相同的 Intended use;且
- 有相同的 Technological characteristics,或
- 雖 Technological characteristics 不同,但沒引起不同的安全與功效問題 (Not raise different questions of safety and effectiveness),且
- 可證明新醫材與 Predicate 一樣安全與有效 (As safe and effective)。
| 與 510(k) 相比,PMA 因為沒有類似品可比對,所以它核准標準雖然一樣是著重在「Safety」和「Effectiveness」,但卻是「自己產生的」利益和「自己造成的」風險、傷害或疾病相比:「a “reasonable assurance of safety and effectiveness” by “weighing any probable benefit to health from the use of the device against any probable risk of injury or illness from such use,” among other relevant factors.」 簡單說,510(k) 是要證明新產品與類似品在利益/風險權衡結果類似後才可上市,但 PMA 及 De Novo 要證明產品利益大於使用風險才可上市。 而「A reasonable assurance of safety」意思是「it can be determined, based upon valid scientific evidence, that the probable benefits … outweigh any probable risks,」;「a reasonable assurance of effectiveness」是指「it can be determined, based upon valid scientific evidence … the use of the device for its intended uses … will provide clinically significant results.」 那什麼是「clinically significant results」?其實就是「Intended use」或「Indications for use」。 以上截錄自〈Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications〉的「A. The Statutory Standard for Safety and Effectiveness」(August 30, 2019 issued)。 上述指引在本文後將縮寫為〈PMD & De Novo B-R assessment guidance〉。 |
因此,了解何謂「Different technological characteristics」很重要:
…the term ‘different technological characteristics’ means, with respect to a device being compared to a predicate device, that there is a significant change in the materials, design, energy source, or other features of the device from those of the predicate device.
Section 513(i)(1)(B) of the FD&C Act (21 USC 360c(i)(1)(B))
2.1.3 何時用到 B-R evaluation?
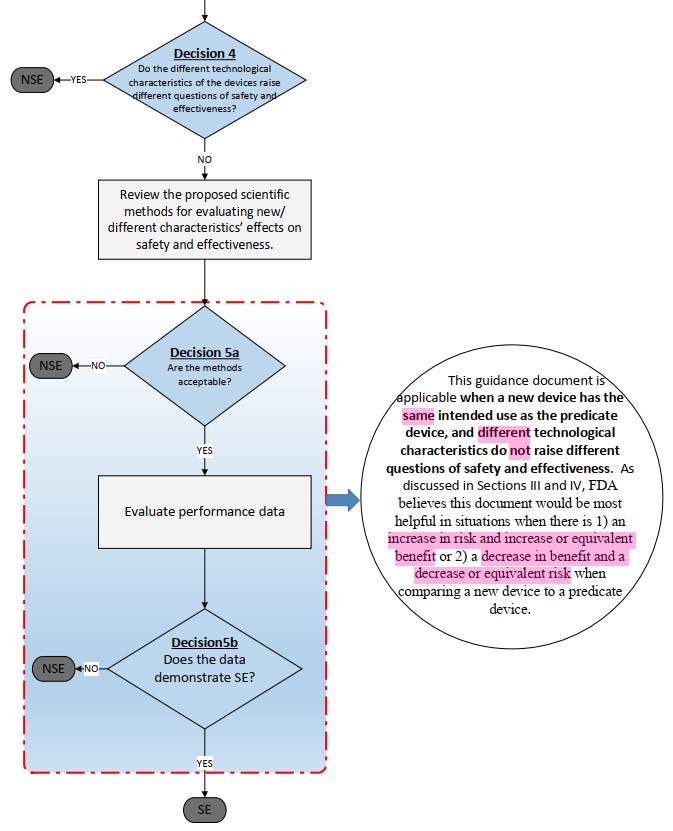
在符合「雖然 Technological characteristics 不同,但沒引起不同的安全與功效問題」後,要進一步評估 Technological characteristics 的差異對於安全與功效的影響,也就是確認「Whether the new device is “as safe and effective” as the predicate device」。
Section 513(a)(2) of the FD&C Act (21 USC 360c(a)(2)(C)) 提到,FDA 透過「weighing any probable benefit to health from the use of the device against any probable risk of injury or illness from such use」來確認產品的安全與功效 (Safety and effectiveness of a device)。
也就是說,在 510(k) Decision-making flowchart 的 Decision 5a ~ 5b 間 (已確認新醫材與 Predicate 的 Intended use 相同,而 Technological characteristics 就算不同,也沒引起不同的安全與功效問題後),可能會動用 B-R evaluation,這也是〈510(k) B-R assessment guidance〉聚焦討論的重點。
將 Benefit 與 Risk 搭配評估,可得以下四句型結果:
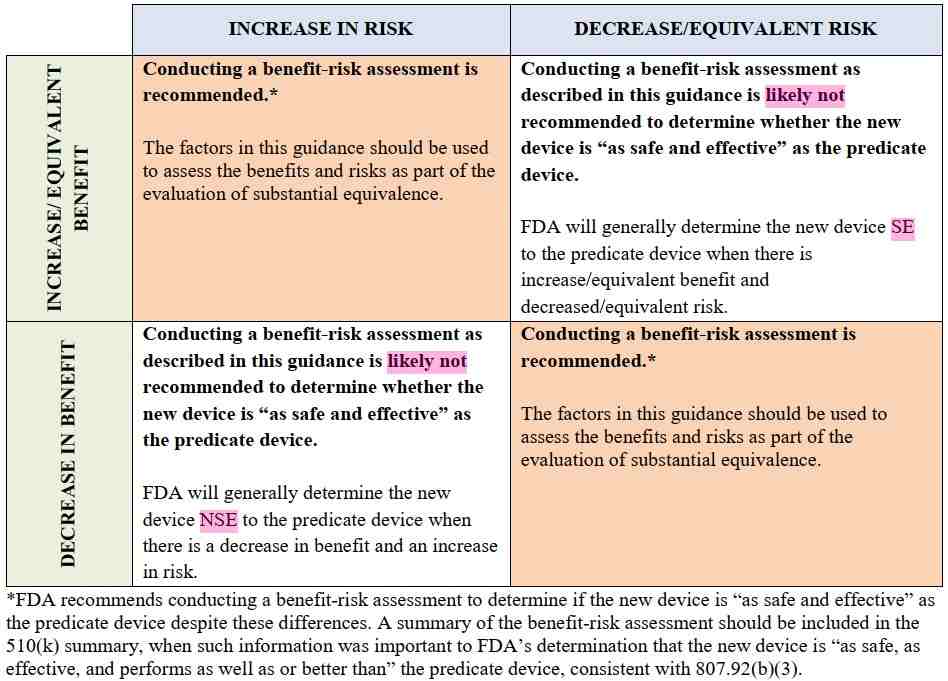
只有在上圖的左上與右下兩個情況發生時,需要 B-R evaluation 來判定新醫材是否「as safe and effective as」Predicate。
| 要特別注意到醫事人員 (Health-care professionals) 和病患的對新醫材利益的重視程度,這些資訊可由「Patient preference information (PPI)」和「Patient-reported outcomes (PROs)」取得。 詳細資訊可參考 FDA 指引〈PPI Guidance〉及〈PRO Guidance〉。 |
所以,只有兩個情況要進行 B-R evaluation,新醫材與 Predicate 比較後:
- Increase in risk & Increase or equivalent benefit,或
- Decrease in benefit & Decrease or equivalent risk。
2.1.4 Benefits 考量點
2.1.4.1 Magnitude
要用科學/臨床方式去評估利益,且特別要注意其程度 (Magnitude) 與形式 (Type) 在不同族群的差異。
一般來說,診斷類醫材的利益多是由準確度 (Accuracy)、再現性 (Reproducibility) 和臨床應用的預期效果來決定。
| 〈PMD & De Novo B-R assessment guidance〉將「Type of benefit(s)」和「Magnitude of the benefit(s)」獨立成兩項,其中: [Type] 1. 可考慮 Clinical management、Patient health、Patient satisfaction 三項,例如,「Improving patient management and quality of life, reducing the probability of death, aiding improvement of patient function, reducing the probability of loss of function, and providing relief from symptoms」。 2. 若為診斷類醫材 (Diagnostics),可從對於民眾健康影響的角度討論利益。例如,「Identify a specific disease and therefore prevent its spread, predict future disease onset, provide earlier diagnosis of diseases, or identify patients more likely to respond to a given therapy.」 [Magnitude] 一般可透過 Scale、Endpoints、Criteria,或者是否有達到預設的健康門檻 (Health threshold) 來評估利益程度高低。 |
2.1.4.2 Probability of the Patient Experiencing one or more Benefits
要評估是哪些族群可受惠,不同族群可能享受到不同利益。
另外,通常要同時考慮「程度 (Magnitude)」及「機率 (Probility)」,因此,要討論是「很大的族群 (Probability) 可接受到一小點利益 (Magnitude)」,或者是「少數族群 (Probability) 可享受到很大的利益 (Magnitude)」。
| 「Magnitude of benefit」和「Probability of benefit」的概念,其實可以類比成風險管理的「Severity of harm」和「Probility of occurrence of harm」,都是要考慮「程度 & 機率」。 |
2.1.4.3 Duration of Benefits
要評估新醫材對病患產生利益的持續時間。
例如,有些是可治癒病患的醫材,有些醫材是屬於病患終其一生都要重複使用的,而後者 (重複使用) 將會比前者導入更多利益或風險給病患,或者後者給病患的利益會隨著重複使用越多次而越來越少。
2.1.4.4 Example of possible Benefits
〈510(k) B-R assessment guidance〉舉出以下範例供大家參考:
- 用更短的時間達到相同的效果 (Reduction in treatment time to achieve same effect);
- 改進機械性能,以降低不良事件發生機率或改善操作方式 (Improvement of mechanical properties to reduce probable likelihood of adverse events or to improve handling);
- 改善臨床的管理、生存機率或病患健康 (例如,改善病患管理、生活品質、增進病患身體機能、避免病患喪失機能、促進症狀緩解),以及病患滿意程度,以上都可由 PROs 量測出來 (Improvements in clinical management, probability of survival, other aspects of patient health status (e.g., effect on patient management and quality of life, improvement of patient function, prevention of loss of function, relief from symptoms), and patient satisfaction in the target population);
- 減少輸出的變異性 (Reduction of variability in device output)。
因診斷類醫材性質有別於治療類的醫材,FDA 另外整理如下:
- 識別特定疾病 (Identification of a specific disease);
- 在疾病的不同階段提供診斷 (Provision of diagnosis at different stages of a disease);
- 預測發病 (Prediction of future disease onset);
- 改善病患操作流程 (Improvement of patient workflow);
- 增進效率 (Increase in efficiency or examination);
- 提供可重複/可定量的結果以優化治療 (Provision of reproducible and quantifiable results contributing to the optimization of therapy and treatment);
- 透過減少漏診/誤診,或識別病患對特定醫療的反應 (伴隨式診斷),以治療疾病,或避免/減少其傳播,以上也都可由 PROs 量測出來 (Improvement of patient outcome (e.g., well-being, health status, safety of patients) by facilitating fewer missed diagnoses (or the right diagnosis the first time, hence the correct treatment plan) and/or identification of patients likely to respond to a given therapy and therefore enable treatment of the disease or reduce/prevent its spread)。
| 〈510(k) B-R assessment guidance〉在「(5) Benefit for the Health-Care Professional, Patient, or Caregiver」中,特別討論「對醫護人員的利益」。 例如,手術器械有更好的人體工學而易於使用 (Surgical instruments with improved ergonomic design for ease of use),或病患監控設備有無線連線能力 (Patient monitoring devices with wireless capabilities) 都可以算是提升 Benefits 的設計。 FDA 說,要證明這類的利益,除了提出佐證資料外,還要說明是用何種可量化的量測方式 (Valid measurement methods) 取得這些資料的。 FDA 再次建議:「Any submitter who is considering developing or presenting valid measurement methods and/or data concerning perspective on benefit for health-care professionals or caregivers in their 510(k) have early interaction with the appropriate FDA review division」。 |
2.1.5 Risks 考量點
本指引〈510(k) B-R assessment guidance〉列出下面多個傷害事件 (Harmful events) 要評估的重點。
2.1.5.1 Severity, Types, Number, and Rates
| 〈PMD & De Novo B-R assessment guidance〉在 Footnote 12 提到,雖然指引將傷害事件型態分成「Severity, types, number and rates」,主要目的是提醒我們在利弊分析時要注意的點,FDA 實際在進行利弊分析時會整體一起看。 |
關於嚴重度 (Severity),分成兩類:
- Device-related serious adverse events:使用此產品導致死亡、傷害、危及生命的疾病,或需要醫療、手術介入以避免永久傷害。(「Serious injury」定義請參考 21 CFR 803.3.(w))
- Device-related non-serious adverse events:由此產品造成的影響,但不符合上述 Serious adverse events 的定義。
而多個傷害事件同時發生 (Multiple harmful events occur at once),可能會讓原本輕微的傷害變成嚴重的結果 (Greater aggregate effect)。
關於類型 (Types),本指引分成下方兩類:
- 產品相關的 (Device-related),以及
- 程序有關的併發症 (Procedure-related complications):由使用此產品「間接」造成的事件,並不歸類於 Device-related (non-)serious adverse events。例如,為了植入醫療器械、或為了收集人體組織樣本而執行的麻醉所產生的併發症 (Anesthetic-related complications)。
| 關於 Procedure-related complications,〈PMD & De Novo B-R assessment guidance〉在 Footnote 14 提到:「These considerations affect the risk profile of in vitro diagnostic devices when the biological material is collected via an invasive procedure for the purpose of performing the diagnostic test.」 所以呀,IVD 不是只有誤診之類的風險喔! |
關於數量 (Numbers)、比率 (Rates):傷害事件的發生比率 (Rate of harmful events),與使用此產品後,
- 在單一病人 (Per patient) 身上所發生的傷害事件數量,或
- 單位時間內 (Per unit of time) 發生的傷害事件數量,有關。
| 〈PMD & De Novo B-R assessment guidance〉在 Footnote 11 定義:「For purposes of this guidance, “rates” means the number of harmful events per patient or number of harmful events per unit of time.」 同時,也將使用醫材所導致的傷害事件的「Severity, types, number and rates」、「Probability」、「Duration」、「Risk from false-positive or false-negative results for diagnostics」歸類於「Extent of the probable risk(s)/harm(s)」。 |
2.1.5.2 Probability
傷害事件的發生機率 (Probability of a Harmful Event) 主要是看「Intended population」中發生傷害事件的機率。
(指引此部分有段話我看不懂:「FDA could factor whether an event occurs once or repeatedly into the measurement of probability.」是要看傷害事件是單一事件,還是有因果關係 (發生一次,造成兩次以上的傷害)?為何指引的「2.1.5.1 Severity, Types, Number, and Rates」就不考慮這點呢?)
| 「Rate」和「Probability」實在很像,我反覆看指引 (+腦補) 後發現兩者差異是:以計算公式來說, 前者 (Rate) 的分母是「『用此產品』的單一病人」或「『用此產品』的單位時間」;後者的分母則是「『預期』會遭遇傷害事件的族群」。 那分子呢?「Rate」的分子是「傷害事件數量」,「Probability」的分子則是「『單位時間』內傷害事件數量」。(好,我知道還是很奇怪…Orz) |
| 在〈510(k) B-R assessment guidance〉的「Probability of a Harmful Event」還有下面一段話: “FDA could factor whether an event occurs once or repeatedly into the measurement of probability.” 但我就不了解,為何在討論「Rate」時不用注意是單一事件或重覆發生?也好奇怎樣材算重覆發生? |
2.1.5.3 Probability of the Patient Experiencing One or More Harmful Event(s)
和 2.1.4.2 Probability of the patient experiencing one or more Benefits 一樣。
2.1.5.4 Duration of Harmful Events
不同類型的傷害有不同「Severity」和「Duration」,兩者要同時考慮。
例如,暫時且較不嚴重的傷害、重複但可逆的傷害,或永久的傷害,而這些不同類型的傷害事件都會在利弊權衡時有不同的考量。
| 〈510(k) B-R assessment guidance〉在「(6) Risk Mitigation」中特別說到,可能有一個新醫材的風險相較於 Predicate 是變高的,但只要有辦法降低風險 (也就是降低傷害事件的發生機率),那也是有機會證明「the new device is “as safe and effective” as the predicate device」。 也就是說,在此比較的風險是「降低後」的風險。 指引中同時也說,常見的降低風險方式有:1) Labeling (E.g., warnings, precautions, contraindications) 或 2) Training。 另外,也可透過 1) 輔助診斷 (Complementary or supplementary diagnostic tests) 或 2) 搭配其他資訊 (E.g., clinical symptoms and family history) 來降低 IVD 的風險。 〈PMD & De Novo B-R assessment guidance〉在「Risk mitigation」(P.14) 中提到,常見降低風險的方法還有「限縮使用」(Restrict the indication to a more limited use)。 |
2.1.5.5 Risk from False-Positive or False-Negative Results for Diagnostic Devices
在此特別將診斷類醫材的偽陽/偽陰性的風險獨立出來。
本指引〈510(k) B-R assessment guidance〉說明,此類醫材的偽陽/偽陰結果可能會:1) 讓病人接受到不必要的治療 (進而招致治療相關的風險)、2) 沒有效診斷出疾病 (因而無法享受到該治療的益處)、或 3) 診斷出錯誤的疾病/病況。
因此,要考慮發生上述情形的可能性 (Likelihood) 與嚴重度 (Severity)。
| 〈PMD & De Novo B-R assessment guidance〉在「B. Assessment of the Risks of Devices」末段特別提到「Severity of their aggregate effect」,可能多個不嚴重的傷害事件同時發生時,會使傷害變嚴重。 |
2.1.6 綜合考量點
除了上兩章節個別關於 Benefits (B) / Risks (R) 的考量外,還要考慮下面這些會增減 B/R 的因子。
2.1.6.1 Uncertainty
找出利益、風險後,還要考慮「不確定性」(換個角度,也可說「確定程度 (Extent of certainty)」) 的影響。如果,證據顯示使用某醫材「可能」有超大利益,但這個「可能」非常地不確定,那…(可能) 還不如證據確鑿 (很肯定) 的小利益。
哪些因素會影響不確定性呢?
例如,實驗設計、實驗執行方式 (員工經驗、量測儀器管理、實驗設計…)、結果分析方式等,都可能提高 B/R 證據的不確定程度。
另一方面,實驗的可重複性 (Repeatability of the study results)、分析方法的確效 (Validation of the analytical approach)、其它類似研究的結果 (Results of other similar studies),以及是否是為此類研究的第一個或為獨立試驗 (Whether the study is the first of its kind or a standalone investigation) 都可能提升 B/R 證據的確定程度 (Level of certainty)。
還要考慮試驗結果對預期療法、使用族群的普遍性 (Generalizability of the trial results to the intended treatment and user population is important)。
像是若一個醫材須操作者有專門技能 (Specialization) 或要先接受大量的訓練 (In-depth user training) 才可使用,則此類臨床試驗結果就可能不是用於廣泛的醫師族群。同樣地,若此醫材預期用來診斷某子群 (Subpopulation),則可能不適用於一般族群。
總的來說,要考慮臨床試驗族群可多大程度地代表預期市場或目標族群 (The degree to which a clinical trial population is representative of the intended marketing or target population)。
2.1.6.2 Characterization of the Disease/Condition
在 510(k) 審查過程中,FDA 還會考量:
- 治療/診斷的疾病與其臨床表現 (Clinical manifestation);
- 疾病如何影響病人的;
- 疾病如何/可否被治療,以及
- 疾病的自然病程 (Condition’s natural history and progression) (例如,是否會自然變好/壞?機率是多少?)
| 〈PMD & De Novo B-R assessment guidance〉在 P.13 提到以下三點會影響 Patient preference information (PPI) : 1. Severity of disease or condition 例如,當面對威脅性命的疾病時,病患通常願意承受多點的治療風險,而如果是診斷嚴重疾病的醫材,使用者通常不太能接受偽陰性的風險。 2. Disease chronicity 若病患已習慣某慢性病,且此病對於病患生活干擾很小,則病患對治療此病的風險接受程度會很低,反之,若某慢性病會長時間使病患越來越衰弱,則病患可能願意承擔較大風險以換取較小的利益。 3. Availability of alternative treatment/diagnostic options 在考慮替代的治療法/診斷法時,須考慮非醫材的治療 (Non-device therapies),並考慮替代方案的「功效」(How effective they are)、「已知風險」(What known risks they pose)、「目前用法」(How they are used in current medical practice)、「利益風險概況」(Benefit-risk profiles)、「如何滿足需求」(How well available alternatives address the needs of patients and providers)。 FDA 在此舉例 (P.14),若某新醫材有一個非常小但重要的利益 (Very small significant benefit),且其利益的不確定性很高,在沒其它替代療法的情況下,此新醫材仍可能獲准上市,因為 FDA 同時還考慮了「The risk to the patient of having no treatment if a device were not approved」。 |
2.1.6.3 Innovative Technology
通常越創新產品,在評估 B/R 時的 Uncertainty 越高。此時,FDA 還會考慮此創新技術對公共衛生 (Public health) 帶來多大的改善。
通常,當產品可能帶來越大的利益,為了讓民眾快點享受到此創新技術的益處,FDA 越可能接受更多的 Uncertainty。
| PMA & De Novo 多是創新到完全沒類似品的情況,因此〈PMD & De Novo B-R assessment guidance〉在「Novel technology addressing unmet medical need」章節還另外提到,FDA 會評估是否「providing a treatment or means of diagnosis where no alternative is available」,這可能會是 FDA 決定准駁 PMA & De Novo 的關鍵,畢竟這類產品常常不會有大量的資料去證明安全與功效,但若核准門檻太嚴苛,又會危害到使用者需求。 |
2.1.6.4 Patient Tolerance for Risk and Perspective on Benefit
不同病患有不同的風險接受程度 (Risk tolerance),這會影響個別病患決定是否承擔風險以換取可能的利益 (Benefit-risk tradeoffs acceptable)。
病患接受程度 (Patient tolerance) 與看待利益的角度 (Perspective on benefit) 是非常主觀的東西,要以病人的角度去看 (Patient-centric assessments),而非以製造廠自己想當然耳的方式武斷下結論。
要如何得知病人對產品的風險、利益的觀點呢?
可透過 PPI study 取得相關資訊,以了解整體利弊 (Overall benefit-risk profile)。例如,了解哪些族群願意承受較大的風險,以取得較多的可能利益、或更多類型的利益,哪些族群比較保守。在做這類評估時,必須考慮病患的「Willingness & Unwillingness」,而非 One-sided evaluations。
Patient preference information (PPI) 定義如下:
The qualitative or quantitative assessment of the relative desirability or acceptability to patients of specified alternatives or choices among outcomes or other attributes that differ among alternative health interventions.
PMD & De Novo B-R assessment guidance | IV. Factors FDA Considers in Making Benefit-Risk Determinations | Patient perspectives
〈PMD & De Novo B-R assessment guidance〉同時也說,當利弊權衡與 Care-partners (e.g., parents) 和 Healthcare professionals 有關時,也會考慮這兩者的好惡 (Preference)。
| FDA 建議,執行 PPI study 前,最好先和 FDA 討論 (Pre-submission)。諮詢方式請參考〈Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program〉。 |
| 〈PMD & De Novo B-R assessment guidance〉在「Patient-centric assessments and patient-reported outcomes (PROs)」章節提到,可透過 Health-related Quality of Life (HRQoL) Measures (健康相關的生活品質量測),以及 Patient-Reported Outcomes (PROs) (病人自述結果) 來達到「Patient-centric metrics」,幫助病患及醫療衛生從業者 (Health care practitioners) 討論治療方式及做決定,也可幫助 FDA 准駁產品上市。 關於 QoL & PRO 的基礎介紹,可參考超清楚的〈病人自述結果在療效評估上的意義和用途〉(家庭醫學與基層醫療|第三十五卷|第六期)。 另可參考 FDA 在 JANUARY 2022 發布的〈Principles for Selecting, Developing, Modifying, and Adapting Patient-Reported Outcome Instruments for Use in Medical Device Evaluation〉。 |
| 〈PMD & De Novo B-R assessment guidance〉在 Footnote 19 對「Unreasonable risk」的解釋真是既白話又貼切: “For the purposes of this guidance “unreasonable risk” refers to a risk that no set of reasonable patients would be willing to endure to achieve a probable benefit.” |
2.1.6.5 Postmarket Data
透過相同類型 (Same type) 產品的上市後資料 (E.g., literature, recalls, registry data, medical device reports),可以進一步了解新產品的利益與風險。例如,確認特定風險是否確實可透過一些方法去降低,或者看看有沒有哪些特殊族群更可能遭受到不良事件。
Section 522 of the FD&C Act (21 U.S.C. § 360l) 以及 21 CFR Part 822.4 授權 FDA 可要求第二、三等級的製造廠執行 Postmarket surveillance。
另外,為了減少製造廠與 FDA 的負擔,也讓有需求的人可以盡早享受到新醫材的利益,所以 Section 513(i)(1)(C) of the FD&C Act (21 U.S.C. § 360c(i)(1)(C) 也授權 FDA 可透過要求製造廠進行上市後管控 (Postmarket controls, e.g., Quality System regulations, postmarket surveillance, and the Medical Device Reporting requirements),以接受更多上市前 Benefit-risk profile 的不確定性 (Uncertainty)。
| 〈PMD & De Novo B-R assessment guidance〉在「Postmarket data」(P.14) 另外還提到,可收集上市後資料來確認降低風險措施的程度與有效性 (Magnitude & effect of mitigations)、是否有辨識出新風險、特定風險是否以被降低。 另外,依 21 CFR 814.82(a),FDA 也有權可在 PMA 核准的同時要求廠商執行核准後研究 (Post-approval studies for PMA)。 |
2.1.7 關於 510(k) 的範例
以下範例聚焦在 Decision #4 之後的 Benefit-risk assessments (更多內容可參考「2.1.3 何時用到 B-R evaluation?」)。
2.1.7.1 Change in Device Design
2.1.7.1.1 背景介紹
有一個新式動力式骨鉗 (Powered rongeur) 在申請 510(k)。
新骨鉗有不同的外型 (Shape),因為這個骨鉗的新外型,510(k) 審查員認為將使得病人暴露出更多的部位 (Exposes additional anatomy of the patient),而可能會有更多的傷害。因此,相較於 Predicate,新骨鉗的風險是增加的。
但是,也因為可接觸到病人的更多部位,所以,新骨鉗可更輕易接觸到以往接觸不到的地方,可加速手術流程 (Expedites the cutting process and minimizes the time needed for surgical procedures)。
製造廠提供性能數據 (Performance data) 來說服,新骨鉗發生審查員設想的那些傷害機率是很低的。另外,也提供動物實驗證明,新骨鉗可加速手術流程。
除此之外,所收集的外科醫生回饋也都說:
…ease of accessing more difficult anatomic areas compared to the predicate device, therefore reducing the likelihood of injury to neighboring tissue when accessing the difficult-to-reach anatomical areas in the patient.
510(k) B-R assessment guidance | V. Examples of Benefit-Risk Evaluation | Example 1
因為新骨鉗有很多不同的尺寸,在動物實驗時也發現,如果用到過大的尺寸,可能會導致增加不良事件的發生機率。
2.1.7.1.2 是否要進行 B-R Assessment?
要,因為這是「風險與利益均增加」的狀況,所以要進行 B-R assessment。
| Benefits | Risks |
|---|---|
| 相較於 Predicate,新骨鉗提供外科醫生一個更易於接觸脊椎附近特殊部位的方法,且這新骨鉗傷害到附近組織的機率更低。 另外,透過動物實驗可了解到,新骨鉗可縮短手術時間,進一步降低麻醉與感染的風險。 | 新骨鉗可能對病人某些部位 (Dura, arteries, veins, and nerve roots) 有更大的傷害機會,但製造廠提供的 Performance data 顯示,發生傷害的機率不高。 不過,動物實驗結果顯示,若用到不適合的尺寸,會增加傷害的風險。 |
額外考量點:
| Risk Mitigation | Benefit for the Health-Care Professional or Caregiver |
|---|---|
| 為避免用到不適當尺寸的新骨鉗,在 Labeling 中說明,如何在手術前量測病人解剖位置,以決定該選擇哪一個尺寸的骨鉗。 | 製造廠提供的資料顯示,外科醫生認為新骨鉗更易於使用,進而降低對病患的風險。 |
SE Analysis:
雖然技術特徵差異 (Technological differences) 顯示,新骨鉗可能會增加 Dura、Arteries、Veins、及 Nerve roots 的傷害風險,但經由下面幾項措施與觀察,
- 動物實驗顯示,發生傷害的風險機率不大,且
- 可縮短手術時間,因而降低手術麻醉與感染風險;
- 透過 Labeling 指導外科醫生挑選適當骨鉗尺寸;
- 外科醫生問券結果顯示,新骨鉗可更容易接近 Predicate 難以接近的脊椎附近特殊位置,進而可減少對病人的傷害,
顯示最終風險可能僅稍高於 Predicate,但同時,新骨鉗的利益也高於 Predicate,「The new device would likely have a comparable benefit-risk profile to the predicate device for the indicated patient population」。
所以,最終 510(k) 判定為 SE。
2.1.7.2 Change in Technology and Possible Change in Principle of Operation
2.1.7.2.1 背景介紹
有一個新的灰指甲 (Toenail fungus, onychomycosis) 低功率雷射治療機,可發射不同於 Predicate 的波長的雷射,所以對 Toenail fungus 會造成不同的光生物效應 (Photo-biological effects),且功率較低。
另外,有別於 Predicate 的脈衝式 (Pulsing sequence) 能量輸出,新治療機能量輸出形式是恆定的 (Constant energy delivery sequence)。
由製造廠提供的 Clinical data (臨床資料) 可知道,新治療機的療效低於 Predicate,但因為新治療機輸出的能量較低,所以有較低的風險。
2.1.7.2.2 是否要進行 B-R Assessment?
要,因為這是「風險與利益均減少」的狀況,所以要進行 B-R assessment。
| Benefits | Risks |
|---|---|
| 新治療機雖提供新的治療方式,但由 Clinical data 可知,療效低於 Predicate。 | 新治療機輸出功率較 Predicate 小,故風險與副作用都較小。另外,也讓使用者配帶雷射防護眼鏡來進一步降低眼睛被雷射傷害的風險。 |
額外考量點:
| Uncertainty |
|---|
| 「療效」是新治療機利益的最大不確定因子,因 Clinical data 結果顯示,成功治癒的比例低於預期。另外,新治療機製造廠所繳交的實驗資料也有不一致的地方。 |
SE Analysis:
因為 Technological characteristics 和 Predicate 有差異,新治療機製造廠因此提交了 Clinical data,想藉此證明與 Predicate 的 SE。
但 Clinical data 結果顯示,新治療機的療效 (也就是 Benefits) 並沒有達到預期效果,且所提交的資料也有明顯的不一致,因此這些 Clinical data 也不太可靠。
雖然新治療機的風險較 Predicate 小,但利益也較小。而且,原本已不大的利益,更因製造廠提供的不可靠資料,提升了 B-R assessment 的不確定性,進一步降低了新治療機的利益。
所以,最終 510(k) 判定為 NSE。
2.1.7.3 New Device with Higher Risk of Malfunction
2.1.7.3.1 背景介紹
有一台新式輸液幫浦 (External infusion pump) 可用在走動、便攜式的情境 (Ambulatory, portable setting)。製造廠宣稱這台新式 Infusion pump 和用於醫院情境 (Hospital setting) 的靜脈輸液/給藥 (Intravenous (IV) delivery of fluid and medications) 的幫浦有實質相等性 (SE)。
與 Predicate 不同的是,此新式幫浦完全是靠電池驅動,使用者介面 (UI) 較簡單,體積也比較小,所以可於救護車、直升機等運輸工具內。因此,若發生 Chemical, biological, radiological, nuclear and explosive (CBRNE) events 時可及時治療病患。
經製造廠的評估發現,新式幫浦的使用者介面足以達成預期用途,且不會增加風險。
但因為可用於移動環境 (Ambulatory environment),所以會有較大機率因外力造成的損壞性故障 (Damage-related malfunction),導致輸送過少/過多液體 (Under-/over-infusion) 或延遲治療。
為了解決上述風險,製造廠透過模擬移動運輸環境 (Ambulatory transport scenarios) 的最壞情況,例如,溫度、濕度、機械力 (衝擊、震動…)、液體滲入 (Fluid ingress)、氣壓海拔等,來測試新式幫浦的耐久性 (Durability)。
實驗結果顯示,在此環境下重複使用的新式幫浦容易有校正偏移 (Calibration drift) 的風險。為降低此風險,製造廠修改 Labeling 提醒使用者常進行預防性維護 (Preventive maintenance)。
2.1.7.3.2 是否要進行 B-R Assessment?
要,因為這是「風險與利益均增加」的狀況,所以要進行 B-R assessment。
| Benefits | Risks |
|---|---|
| 新式幫浦的小巧外型,可使醫事人員在侷促的運輸環境下進行靜脈注射 (Intravenous (IV) therapy)。 此外,製造廠也透過模擬測試證明新式幫浦可用於多變的溫度與濕度,所以也適用於 CBRNE events。 | 在移動環境下使用新式幫浦會增加校正偏移的風險,因而導致輸出過多/少的液體。 從實驗結果來看,只有在反覆於移動環境 (Ambulatory environment) 使用幫浦會發生校正偏移的問題。 |
額外考量點:
| Benefit for the Health-Care Professional or Caregiver | Risk Mitigation |
|---|---|
| 相較於 Predicate,新式幫浦可讓醫護人員將靜脈治療的場域,從醫院延伸到移動環境中,從而滿足在那情境下的病患緊急需求。 | 修改 Labeling,告知使用者每使用 100 小時要進行預防性維護,以避免校正偏移。此頻率高於 Predicate 所建議的。 |
SE Analysis:
有別於 Predicate,新式幫浦的小巧、簡化 UI、電池驅動的特性,使其可適用於移動的情境 (Mobile ambulatory setting),進而提供移送中的病人 (Patients in transit),或突發公衛事件中的大量患者 (Mass number of patients in a public health emergency) 治療。
另外,從製造廠的非臨床性能數據 (Non-clinical performance data) 得知,隨之而來的風險就是,這種特殊的使用環境容易導致校正偏移,造成輸送過少/過多液體 (Under-/over-infusion) 或延遲治療。
為了降低風險,製造廠修改 Labeling,提高了建議預防性維護頻率。
所以,最終 510(k) 判定為 NSE。
2.1.7.4 Material Differences Resulting in Different Device Performance
2.1.7.4.1 背景介紹
有家製造廠研發出合成橡膠乳膠的男性保險套,並以傳統的天然橡膠乳膠 (Natural rubber latex) 保險套作為 Predicate。兩者之間的技術特徵差異只有「材質」:合成 vs. 天然。
對於合成材質保險套的主要疑慮是,性交時可能較天然材質保險套更容易破損 (Breakage) 和滑脫 (Slippage)。
製造廠希望透過臨床研究去證明 (痾~這個臨床研究真害羞~~),合成材質不會比天然材質差 (Non-inferiority)。後來,研究結果都有達到原先設定的主要指標 (Primary endpoint),但滑脫率 (Slippage rate) 稍高於天然材質保險套。
2.1.7.4.2 是否要進行 B-R Assessment?
要,因為這是「風險與利益均增加」的狀況,所以要進行 B-R assessment。
| Benefits | Risks |
|---|---|
| 新材質保險套特別可利益到原先對天然材質保險套過敏的使用者。 | 在性交過程中,新材質較易滑脫進而增加非預期的懷孕機率,以及性傳染病 (Sexually-transmitted infections, STIs) 的傳播機率。 |
額外考量點:
| Risk Mitigation |
|---|
| 為了降低新材質保險套略高的滑脫率 (Slightly higher slippage rate),製造廠新增 Labeling 說明,僅有對天然橡膠乳膠過敏的人才可以用此合成材質保險套。 |
SE Analysis:
雖然新材質保險套有略高的滑脫率,但卻提供了對天然材質過敏的使用者新的避孕 (Contraception) 與預防疾病 (Prophylaxis) 的方式。此外,製造廠透過 Labeling 降低因滑脫而造成的風險。
所以,最終 510(k) 判定為 NSE。
2.1.7.5 Different Principle of Operation Used to Achieve Same Therapeutic Outcome
2.1.7.5.1 背景介紹
相較於可達相同預期療效的 Predicate,此新醫材可對嘴巴輸出更高的壓力,並以不同的作用原理 (Principle of operation),去治療成人的阻塞性睡眠呼吸中止 (Obstructive Sleep Apnea, OSA)。
疑慮是,新的作用原理可能會部分閉鎖口腔,進而限制使用者用鼻子呼吸。
製造廠用測試報告 (Bench test) 說明,並不會因為較高的壓力,而讓使用者在鼻腔阻塞 (Nasal obstruction) 的同時,閉鎖其嘴巴。
另外一個 28 天的研究報告結果顯示,新醫材可降低呼吸中止指數 (Apnea-hypopnea index, AHI),但降低幅度沒有 Predicate 多。
而且,因為較高的壓力,相較於 Predicate,新醫材的病患依從性 (Patient compliance) 降低、併發症機率 (Complication rate) 變高。
2.1.7.5.2 是否要進行 B-R Assessment?
不用。因為相較於 Predicate,新醫材的風險增加,且利益降低,一般來說,FDA 會判定為 NSE。
2.1.7.6 Comparative Testing Resulting in Substantially Different Results
2.1.7.6.1 背景介紹
兩套產品都可用來量測凝血脢原時間 (Prothrombin time, PT) 國際標準化比值 (International normalized ratio, INR) (可稱為「ProTime INR」) 及凝血因子量 (Coagulation factor levels),但相較於 Predicate 是從動物組織中萃取而得到的成分,新產品採用重組 DNA 技術來取得所需要的成分。
雖然新產品在低量凝血因子情況下,血塊形成 (Clot formation) 時間較 Predicate 長,但校正後,此二產品在 Fibrinogen、Coagulation factors II、V、VII、X 的 INR 有強相關 (Strong correlation)。
另外,新產品的精確度 (Precision),以及不同天 (Inter-day)、批次間 (Inter-lot) 的差異比 Predicate 有更高的再現性,且凝血脢原干擾因子 (PT interferents) 對新產品並沒有非預期的影響 (No unexpected effect)。
檢視相關 Postmarket medical device reports (MDRs),僅有一款重組 DNA 技術的產品被抗生素干擾,而此抗生素在此之前沒有被發現與凝血脢原干擾源 (PT interferents) 有關。製造廠的實驗顯示,新產品並不會被此類抗生素干擾。
| 「ProTime INR」的相關知識可參考〈一文搞定凝血從理論到臨床應用〉。 |
2.1.7.6.2 是否要進行 B-R Assessment?
不用。新醫材和 Predicate 有相同的風險,但利益卻變多。一般來說,FDA 會判定為 SE。
2.2 關於 PMA & De Novo Request 的 B-R Evaluation
CDE 在 2013 年 有整理舊版〈PMD & De Novo B-R assessment guidance〉的重點,大家可參考看看〈醫療器材上市前審查考量之利益與風險權衡要素〉。
2.2.1 前言 (不是廢話)
這部分是參考於 AUGUST 2019 發行的〈Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications〉(後面簡寫為〈PMD & De Novo B-R assessment guidance〉) 而成的筆記。
不同於 510(k) 只要證明 SE 即可上市,FD&C Act Section 513(a) (21 USC 360c) 要求 PMA 的申請者,除了 FDA QSR (21 CFR Part 820) 外:(De Novo Request 類似於 PMA)
- 須透過「Weighing any probable benefit to health from the use of the device against any probable risk of injury or illness from such use」,
- 以證明「Reasonable assurance of safety and effectiveness」,以及
- 「The device will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling of the device」(Section 513(a)(3)(A) of the FD&C Act) 來取得上市許可。
2.2.2 Benefits/Risks 考量點
因為〈PMD & De Novo B-R assessment guidance〉在此部分非常大程度地與〈510(k) B-R assessment guidance〉類似,因此我將此部分穿插於 2.1.4 Benefits 考量點、2.1.5 Risks 考量點、2.1.6 綜合考量點中。
2.2.3 關於 PMA/De Novo 的虛構範例
可參考 CDE 2014 年的〈醫療器材上市前審查考量之利益及風險權衡要素〉中所整理的一個範例。
2.2.3.1 Example 1
2.2.3.1.1 背景介紹
某植入式醫材可用來治療無其它替代療法的嚴重慢性疾病。該醫材已完成臨床試驗,此試驗中,只有一半的受試者會啟動植入的醫材。
此試驗的主要指標 (Primary endpoint) 是利益的程度 (Magnitude of the benefit)。例如,相較於傳統照護,此醫材可降低多少受試者的症狀。
2.2.3.1.2 利弊權衡
| Benefits | Risks |
|---|---|
| 1. 75% 受試者體驗到明顯好處 (Substantial benefit),符合預設的主要指標。 2. 一般來說,維持良好活動力的病患有較長的預期壽命。 3. 無法估計利益持續時間 (Duration of the benefit),因臨床試驗只有一年。 | 1. 傷害事件發生機率小於 3%。 2. 植入手術自有其風險,此手術非常規手術,文獻統計約有 1% 的死亡機率。 3. 永久植入會帶來更多風險,且可能難以移除。例如,即使關閉本產品,也會有斷裂、機械故障或不良生物反應。 |
| Uncertainty | 1. 很難確定受試者得到的好處,是器材還是手術造成的。 2. 因臨床試驗只有一年很難確認利益是否超過一年。 3. 植入後,只有 75% 的受試者會完全成功 (得到好處)。 |
| Patient Perspectives | 即使只有 75% 可得到好處,但大部分病患都願意承擔植入此器材的風險,因為這個病症很嚴重,且沒其它替代選擇。 |
| Risk Mitigation | 植入及移除 (Explant, if necessary) 本醫材的手術危險的,但可藉由專門訓練的外科醫生執行手術來降低風險。 |
2.2.3.1.3 准駁考量
有滿高比例 (75%) 受試者可由此器材獲得利益。
雖然永久植入此醫材會暴露病人於顯著風險 (Significant risks) 中,且仍有一些不確定性無法由臨床試驗確認。然而,從中獲益的受試者在症狀緩解 (Symptom relief)、改善生活品質方面卻非常顯著,且大部分病患願意承受此器材的風險以換取相關利益。
另外,雖然風險大,但可透過訓練有素的專門外科醫生執行手術來降低風險。最後,此嚴重慢性病沒有替代療法。
因此,FDA 可能會核准此產品。
2.2.3.2 Example 2
2.2.3.2.1 背景介紹
某醫材可利用置換病患的記憶來治療 Alzheimer’s disease、Dementia 以及其它記憶力相關的疾病。此醫材設計成永久植入,且為了讓其順利運作,病患須接受腦部切除術 (Brain resection)。本產品透過下載病患的記憶到電腦晶片上來達到其功能,一旦植入本產品,病患將無法使用任何殘餘記憶。
2.2.3.2.2 利弊權衡
| Benefits | Risks |
|---|---|
| 1. 可顯著改善早期癡呆症 (early stages of dementia)。 2. 只可稍微改善 (Minimal improvement) 更晚期 (More advanced stages) 的患者。 | 1. 就算一般僅由訓練有素的神經快科醫師執行植入手術,手術風險仍然很高 (8% 手術會造成 Serious adverse events)。 2. 嚴重不良事件包含:Partial paralysis、Loss of vision、Loss of motor skills、Vertigo、Insomnia (預測機率 1%)。 3. 非嚴重不良事件包含:Temporary personality shifts、Mood swings、Slurred speech (預測機率 5%)。 |
| Uncertainty | 1. 只有極少數受試者 (Number of subjects eligible)。 2. 但試驗設計與進行均很好。 3. 結果是可靠 (Robust) 且具普遍性 (Generalizable)。 4. 結果顯示,記憶喪失初期的受試者可獲得最好的試驗結果。 |
| Patient Perspectives | 1. 因為此類病對患者生活品質 (QoL) 影響很大,故患者以及其照護者 (Care-partners) 通常願意承受非常高的風險以換取改善病狀、減輕生病後期照護負擔的機會。 2. 較老年紀的患者可能較不願意承受相關風險。 3. 較晚期的患者由此醫材得到利益的機率較低,且因為此病的關係,較難評估此類病人的風險承受度。 |
| Availability of Alternative Treatments or Diagnostics | 目前沒有替代療法 |
| Risk Mitigation | 1. 限於由接受過專業訓練的外科醫師使用 (但性格轉變的風險仍無法降低與遇測)。 2. 透過 Labeling 接露,有嚴重病徵的病人較難由此器材獲得利益,並指示本產品用於最有可能獲得利益的初期病人。 |
| Novel Technology Addressing Unmet Medical Need | 1. 目前沒有類似科技,也沒其他療法可提供目標族群這種程度的利益。 2. 未來此產品的改善有機會治療其他影響認知功能的疾病。 |
2.2.3.2.3 准駁考量
可明確知道本產品會給哪個族群顯著利益,也可知道哪個族群使用只會有很少的利益。即使臨床試驗樣本數很少,但試驗結果的品質很好,所以信賴區間 (Confidence intervals) 很窄。
而不確定性來自於用樣本數推估整個預期族群,這種不確定是用這種方法本來就常見的 (The uncertainty about results is the usual uncertainty resulting from drawing inferences from a sample in the study to the population in the market)。
本產品風險很大,但可透過訓練有素的外科醫師執行植入以降低部分風險。
為了要獲得本產品的最大利益需要於疾病早期植入本產品,本產品將會很長時間留置於體內,因此暴露於風險的時間很長。所以,獲得最多利益的病患也將承受最大風險。
廠商提供的資料顯示,許多有記憶障礙 (Memory disorders) 的病患願意嘗試這個有顯著風險的創新療法,以保留他們的記憶與生活品質 (QoL)。
目前沒有其他的替代療法。儘管本產品有很高的風險,但因為可能帶來的利益,以及此疾病不可治癒的自然進程,所以還是有病患可接受。而且,風險是已知且可量化的。
最終,FDA 可能會核准此產品,但需在 Labeling 中永久標示 8% 的嚴重不良事件發生率,以及限制本產品只可由受過高度訓練的外科醫師植入。
2.2.3.3 Example 3
2.2.3.3.1 背景介紹
有一新型血清 In vitro diagnostic device (IVD) 可將診斷為 BI-RADS 4 的乳房攝影結果分成兩類:
- 低癌症機率族群:此類病患確診為乳癌的機率較低,因此可再觀察數月,避免切片 (Biopsy) 相關問題 (Morbidity);
- 其它族群:依循現有的標準指引立即切片化驗。
此 IVD 預計的 Intended use 為:
The in vitro diagnostic test measures 10 peptide analytes and yields a single qualitative result. The test is intended for females 40 years or older following mammography of a breast lesion with a BI-RADS of 4 result to aid physicians in the decision to recommend a breast biopsy.
Negative test result (Low Risk): immediate biopsy is not recommended, wait a few months for further tests.
Positive test result (High Risk): immediate biopsy is recommended.
臨床試驗結果如下:

2.2.3.3.2 利弊權衡
| Benefits | Risks |
|---|---|
| 使 57% (228/400) 受檢者免於立即切片所造成的可能風險。 | 偽陰性比例 1.3% (3/228 = 1 – NPV):本產品判斷為低癌症風險的 BI-RADS 4 受試者,其切片結果顯示為癌症的比例。 也就是說,若未來這產品上市,將有約 1.3% 的病人因為沒有立即切片檢查而延誤癌症治療。 為此,廠商解釋,在非切片檢查的 BI-RAD 4 受試者中 (Non-biopsied BI-RADS 4 subjects),臨床上可接受的癌症患病率 (Clinically acceptable prevalence) ≤ 2%。理由是: a) BI-RAD 3 病患通常建議不要立即切片檢查;而且 b) BI-RADS 3 病患的預期乳癌患病率為 2%。 另外,從臨床研究結果發現,可量測利益風險機率 (Benefit-risk odds) 為 75 (225/3),且未立即切片檢查的 BI-RADS 4 受試者的預期風險比 BI-RADS 3 病患低。 |
| Uncertainty | 利益風險機率 (Benefit-risk odds) 並非針對「避免切片後遺症 (Morbidity) 的利益」和「錯失切片可檢出癌症的風險」兩者的比較,也就是說,Benefit-risk odds 中的利益和風險兩者類型不一致。 另外,也無法確定 BI-RADS 4 中漏診的乳癌在臨床上造成的影響和 BI-RADS 3 相等。 |
| Patient Perspectives | 一般來說,病患對乳癌的延誤診斷與治療接受度較低,需要和避免切片的相關問題 (Morbidity) 的利益比較。 |
| Availability of Alternative Treatments or Diagnostics | 目前沒有其他類似預期用途的 IVD 產品被核准 (Cleared or approved)。 |
| Risk Mitigation | 所有檢查結果為陰性的女性都須接受後續進一步地追蹤與檢驗。 |
2.2.3.3.3 准駁考量
利益與風險的種類與機率已有合理地定義出來。也已提出臨床實務 (Clinical practice) 上的風險可接受度,並與測試結果一致。
但因利益/風險兩者用來比較的類型不同,所以要有更多的資訊以決定如此的取捨 (Trade-offs) 是否可接受。
鑒於利益尚未確定,且風險 (對非常小眾的人) 可能非常大,FDA 可能不會核准此產品,但可能會先送交給諮詢小組 (Advisory panel) 評估後再決定。
2.2.3.4 Example 4 — De Novo
2.2.3.4.1 背景介紹
有一新的獨立治療醫材 (Standalone therapeutic device) 可提升侵入式、高風險植入式醫材的穩定性。此新醫材可輔助主醫材 (Primary device) 的植入,或可用來輔助已植入、正在運作的主醫材。
有一項針對此新醫材的前瞻 (Prospective)、多中心的單臂 (Single-arm) 臨床試驗,受試者有 200 人。此臨床試驗的主要指標 (Primary endpoint) 是利益的程度 (Magnitude) ,例如,相較於沒有輔助,在有此新醫材輔助的情況下,使主醫材免於移動與故障的情況效果如何。
2.2.3.4.2 利弊權衡
| Benefits | Risks |
|---|---|
| 經過一年的追蹤,沒有受試者所植入的醫材有移動,且只有兩位受試者有發生器材故障相關的併發症。相較於單獨植入主醫材,搭配此新醫材植入可顯著的改善病醫材植入後的移動情況。 | 經過一年的追蹤,沒有主醫材斷裂,且只有少數 (Handful) 的輔助系統 (此新醫材) 故障,沒有任何 Serious adverse events。輔助系統 (此新醫材) 故障的風險並不高,因為就算故障,也不太可能導致主醫材的全面失效。 儘管所有植入式醫材的手術自有其風險 (例如,1% 的手術致死率),但此新醫材是與主醫材一同植入,不用承擔額外的手術風險。或者,若植入新醫材是為了提升故障的主醫材,那也是原本就要動手術去修復故障的主醫材。 因此,依資料來看,透過手術植入新醫材並不會增加病患的風險。 FDA 確認此輔助系統 (新醫材) 屬於中低風險 (Low-to-moderate risk),相關風險已明確了解,且風險可透過 General controls 與 Special controls 來降低。(另可參考「什麼是一般控制(General control)和特殊控制(Special control)」) 因此,本新醫材適用於 De Novo 上市途徑。 |
| Uncertainty | 此臨床試驗僅追蹤一年。針對永久植入醫材,更長的追蹤時間可降低長期使用的安全功效的不確定性。 |
| Patient Perspectives | 使用此新醫材 (輔助系統) 的病患,要嘛就是已經歷手術植入主醫材,要嘛就是因為主醫材而導致併發症,而此新醫材可透過非手術的方式來矯正這些併發症。 臨床試驗結果顯示,使用新醫材後,未來的病患會因主醫材更好的穩定性而受益。因此,大部分病患都說,他們可能願意承受可能的風險已獲取可能的利益。 |
| Risk Mitigation | FDA 建立 Special controls 來降低相關風險。 例如,Biocompatibility、Sterility、Safety and effectiveness data (包含 Clinical performance data、Durability、Compatibility、Migration, Resistance、Corrosion resistance、Delivery、Deployment)、MR-compatibility 評估、EMC 的確效、限制此產品為處方用 (Prescription use),並於 Labeling 明確說明相關的安全與功效。 因為此新醫材不用額外的植入手術,因此手術風險不是問題。 |
| Novel Technology Addressing Unmet Medical Need | 此醫材是第一個可修復有問題主醫材的系統,給醫生一個重新固定位置不正確、移位的主醫材的微創選項 (Minimally-invasive option)。 |
2.2.3.4.3 准駁考量
臨床試驗結果保障了至少一年的臨床功效。且更重要地,本產品僅是支持與補充其他醫材的功效,此產品的故障並不會顯著地影響到主產品的表現。本產品帶來的風險並沒有到達 Class III 的水平。
所有產品失效相關的風險都可以透過 Special controls 去處理。
鑒於本產品的利益、透過 Special controls 降低風險的能力,且本產品非用來支持生命 (Life supporting) 或維持生命 (Life sustaining),因此 FDA 可能會批准 De Novo 請求,將此產品歸類為 Class II。
2.2.4 關於 PMA/De Novo 的真實案例
2.2.4.1 癌症治療
一個治療非常罕見癌症的產品,其表現雖有一些不確定性,但可和標準療法差不多 (不過也沒比較好)。然而,用此產品並不會像其餘標準的抗癌療法那樣會造成許多傷害 (Harmful effects),且無論新產品或標準療法,兩者都無法治癒癌症 (Neither treatment was curative)。
這癌症期程發展很快,所以參與者被確診後只有很少存活時間。
FDA 核准此產品,因為此產品功效和現行標準療法差不多 (雖然有些不確定性),但不會造成如標準療法那樣的嚴重副作用。
2.2.4.2 永久植入式心血管監測儀
有一個永久植入的新心血管監測裝置預期用來診斷心臟衰竭 (Heart failure)。相關研究結果顯示,此裝置可減少心臟衰竭病患住院天數約三天。然而,此裝置的植入手術需住院兩天。
市面上現有類似產品可給予與新產品類似程度的利益,但卻不用植入手術。
FDA 判定,省下一天住院時間的利益並沒有超過超過手術併發症的風險,因此不核准此產品。
2.2.4.3 永久避孕器
一個新的永久避孕器可利用專門的輸送導管透過陰道放置在生殖系統。此產品是永久植入,且不預期要移除。植入 (Explantation) 此產品需要手術。
臨床資料顯示,此產品兩年內的避孕效果是有效的,且只有低機率會有不良事件。
但是研究發現,在某些案例中,醫生難以正確地放置此產品,且也在某些受試者的後續 X 光追蹤中發現此產品斷裂。
鑒於本產品長期效應 (Long-term impact) 的不確定性,以及斷裂的可能 (並沒在任何 Bench and animal testing 中發現此問題),還有替代療法的安全與功效,FDA 認為此產品並不可核准給預期病人族群使用。
2.2.4.4 植入式醫材
相較於標準療法,有一個獨特設計的植入式醫材也可用來處理類似的症狀。雖然現行的標準照護方式很好,但會限制病患的移動性,而此新醫材則不會影響。
根據臨床試驗的功效相關資料顯示,相較於現行標準照護方式,新產品可顯著改善功能性結果 (Functional outcomes)。
然而從安全方面來說,新產品呈現出不同於現行標準照護方式的不良事件。這風險可透過外科人員培訓,以及適當的 Labeling 來降低。若隨著時間推移而本產品出現故障,醫生也可轉向採用現行標準照護方式。
即使有不同的不良事件,此產品可能的利益還是高於風險,因此 FDA 核准此產品。
2.2.5 FDA 利弊評估表
在〈PMD & De Novo B-R assessment guidance〉的 Appendix B 提供了 FDA 審查員執行利弊權衡時的參考評估表。另外,指引的 Appendix C 也用此表去評估 本指引 2.2.3 章節中關於 PMA/De Novo 的虛構範例。
非常建議大家去看看 Appendix B 及 Appendix C!因為評估表列出很多評估重點,可幫助製造廠系統性地完整評估利益與風險。
Appendix B 的評估表大致架構如下:
2.2.5.1 Assessment of Benefit
「Is there any evidence of clinical benefit?」是開頭第一項問題,若連這項都是「No」,那整個申請就被駁回了。
第二個問題是「What is the extent of uncertainty for the benefits? 」
2.2.5.2 Assessment of Risk
類似評估利益的問題,這部分兩個主要問題是「Are known/probable risks more than minimal?」及「What is the extent of uncertainty for the risks?」
2.2.5.3 Assessment of Benefit-Risk
FDA 在此特別說明,利益與風險權衡僅是 PMA 或 De Novo 准駁的考量點之一,還有許多其它要求 (例如,品質管理系統) 須符合才可准許上市。以下是此部分的評估問題:
- Do the Benefits outweigh the Risks, considering the assessment of Benefit and Risk and the extent of uncertainty identified above? (若還無法確認,則須接著進行以下的評估)
- Do the Benefits outweigh the Risks, when taking into account the following additional considerations?
- Can the risks be mitigated, so that Benefits outweigh the Risks? Consider if the Benefits outweigh the Risks if risk mitigation strategies are incorporated to lower the probability of a harmful event occurring and improve the benefit-risk profile of the device.
- Do the Benefits outweigh the Risks considering the use of postmarket actions?
- Is there any evidence of clinical benefit for a modified Indications for Use?
3 留言
搭版問超強大神有相關經驗嗎?
客戶要拿自家產品去巴拿馬註冊
其中需要我們(製造商)提供產品的”風險分析和風險降低措施”(文字是risk analysis and risk reduction measures)
看文字是不需要完整的風險管理報告
目前手邊有完整的風管報告
但是不知道重點要怎麼呈現,求救T_T
或是有可以參考與巴拿馬醫材申請相關的資訊可以提供:)
他們可能只要看 FMEA 表 (Risk analysis -> Risk evaluation -> Risk control),不用看整個計畫,所以不用繳交前面的產品描述、預期用途,也不用給後面的 Production/Post-production activities 等等 (?)
感謝回答!!
結果風管文件如您所說, 只需要簡要的東西~~